


How federal research continues failing people with myalgic encephalomyelitis, part I
The long and twisted history of psychologizing myalgic encephalomyelitis and post-acute infection syndromes

Welcome back to the breakdown! Today we’re veering slightly off topic to cover a recent article published in the journal Nature Communications that has already garnered significant controversy within patient circles. The paper by Walitt et al1, entitled “Deep phenotyping of post-infectious myalgic encephalomyelitis/chronic fatigue syndrome”, is the culmination of 8 years of work stemming from a 2016 initiative by the National Institute of Health (NIH) to study the clinical presentation of myalgic encephalomyelitis (ME)2, a neuroimmune disorder that typically develops following an infection or immune trigger.3 ME is a criminally understudied disease with no clinically validated treatments or cures; the hallmark symptom, post-exertional malaise (PEM), involves a flu-like symptom crash following low levels of exertion, a highly disabling symptom that can dramatically impair quality of life. A significant proportion of long COVID cases have a similar presentation, and early research has shown overlapping physiological mechanisms between these subgroups.4 Despite the large and growing public health burden presented by ME, it remains one of the most under-funded diseases relative to its disease burden in the United States.5
I was excited to see an email in the early morning hours of Wednesday Feb 21st announcing the hallmark publication from the NIH’s first major investment in ME research, but as I read through the manuscript I encountered the all-too-familiar framing of ME as a disease of nonspecific fatigue rather than a neurological disorder characterized by an abnormal response to exertion. While the study does contain new discoveries about the clinical presentation of ME that will contribute to research, it also contains some major design, analytical, and interpretational flaws that were not addressed despite going through peer review; even in the best-case scenario, the study was still designed and written in a way that can blow back on ME patients. From an outside perspective it may be difficult to understand why patients and advocates are so upset by this study, so in this post I hope to outline the historical context that precipitated this latest wave of disappointment in ME research. Stay tuned for Part II, in which I will break down the manuscript itself in great detail. Let’s dive into it.
TL/DR: the breakdown
Research about myalgic encephalomyelitis dates back to the early 20th century, when at least 14 major outbreaks of a mysterious neurological disorder occurred around the world. At first it was thought to represent an unusual manifestation of paralytic polio.
ME was classified as a neurological disorder by the WHO in 1969 and was immediately met by pushback from a group of doctors who insisted that mass hysteria was the real culprit. The major rationale for this was that the disease mainly impacted women.
In the 1980s, three major outbreaks in the US and the UK drew the attention of health agencies and officials. Doctors treating these patients noted many had antibodies suggestive of recent infection or reactivation of the common viral pathogen Epstein-Barr Virus (EBV).
Officials at the CDC conducted a small trial measuring the impact of the antiviral agent acyclovir on symptoms and basic blood markers. Acyclovir failed to outperform placebo, although the majority of their physiological data was omitted from the publication. Many researchers incorrectly interpreted the negative results in this study to mean that the disease had no biological drivers.
Around the same time, officials at the NIH and CDC convened a panel of “expert” physicians who rebranded the disease as chronic fatigue syndrome (CFS) and significantly changed the diagnostic criteria to focus on medically unexplained fatigue. Officials like Dr. Stephen Straus would go on record to state that CFS was not a specific disease but instead the result of unstable personalities or stress from unrealistic expectations.
A group of psychiatrists in the UK had similar beliefs about the nature of CFS and began promoting the “biopsychosocial” model of the syndrome, which posits that CFS is driven by physical deconditioning and chronic activity avoidance. Essentially, they believe that patients try to return to baseline levels of exertion too quickly following an acute illness, which causes them to feel worse. This results in patients avoiding exercise, leading to further physical deconditioning and reinforcing “unhelpful” cognitions about exercise causing symptoms.
One of the largest interventional trials for CFS was conducted in the UK testing the impact of graded exercise therapy (GET) and cognitive-behavioral therapy (CBT) versus what they called adaptive pacing or standard medical care. The study authors claimed the GET+CBT approach led to moderate improvements in self-reported fatigue and physical function, but critics quickly highlighted a variety of methodological and ethical issues. The study was largely debunked.
Despite low rates of funding, more and more evidence has accumulated showing that ME is a biological disorder. This includes replicated findings that people with ME exhibit significant deterioration in performance and capacity on the second day of a two-day cardiopulmonary exercise test, whereas controls and people with other conditions repeat or even exceed their previous performance. Other studies have shown marked changes in gene expression following exercise, and demonstrated sustained neuroinflammation in people with ME.
In 2015 the NIH announced its plan to launch a integrative cross-institutional study to deeply profile people with ME, although advocates quickly noted potential conflicts of interest and points of concern with the proposed studies. Part II will cover the study itself in more detail.
The psychologization of myalgic encephalomyelitis
Despite the continued lack of interest by the biomedical research community, the syndrome known as ME has been described for decades, originating from a series of outbreaks of unexplained illnesses across the world in the early 1900s. Between 1930 and 1955, there were at least 14 major outbreaks of a mysterious neurological disorder that caused long-term illness in a substantial percentage of affected patients. The outbreaks were widely distributed across continents; at the time, physicians suspected some type of atypical polio that was primarily spreading among hospital staff. The condition was given several names including neuromyasthenia and Iceland Disease before settling on ‘benign myalgic encephalomyelitis’, so named due to the high prevalence of muscular pain (myalgia), central nervous system manifestations including severe headaches and neck pain, and evidence of immune dysfunction (lymphadenopathy, low grade fever).
At this point, I think it’s important to recall that all diseases and disorders of physiology are socially constructed; that is to say, concepts like discrete disease states are categories defined by humans and applied to observable phenomenon. Diseases can and do wreak havoc on the body well before they are recognized or named by the medical establishment, a pattern well illustrated by what we now call migraine auras. Classical migraine auras are believed to originate from a neurological phenomenon known as spreading depression, where a group of misfiring neurons in the visual cortex essentially run out of fuel and enter a quiescent recovery phase while their aberrant activity spreads to the surrounding cells, resulting in a visual disturbance that spreads across the field of vision over a span of several minutes. Most of this was worked out in the late 20th century using modern electrophysiology and later neuroimaging techniques, but migraine auras (or scintillating scotomas if you’re feeling fancy) have been accurately described for centuries, long before we understood the way the brain processes visual information. The visual disturbances were often described as hallucinations, although we now understand it to be mechanistically distinct from hallucinations induced by psychosis or psychedelic drugs. The doctor Hubert Airy sketched out his hallucinations in the late 19th century, an image that many migraine sufferers can immediately identify as a perfect portrayal of our auras.
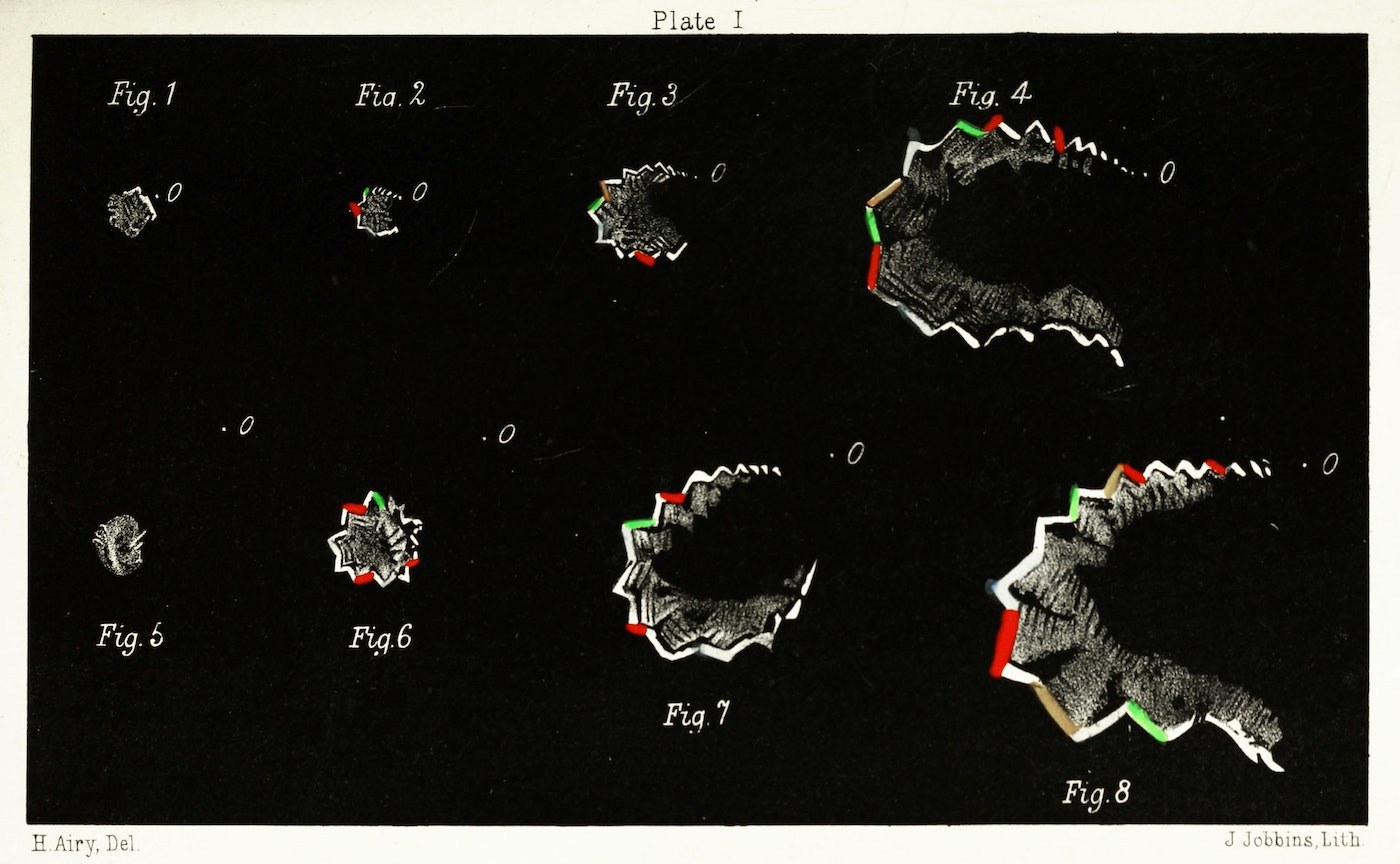
Dr. Airy and others with these auras experienced classical migraines even though that term was not created or used during their lifetimes. Instead, it took centuries worth of scientific progress for us to convince ourselves that migraine auras were not simply hallucinations or hysteria, mainly because we developed techniques capable of measuring the underlying phenomenon in “objective” terms. A relatively common neurological disorder was misunderstood for hundreds of years due to a major false assumption: that the absence of evidence of a physiological disorder is equivalent to evidence of absence of a biological cause. In other words, clinicians historically tend to interpret a handful of “normal” test results to mean you cannot possibly have any kind of organic disease, despite the inherent implausibility of systematically ruling out every disease known to man through a simple blood test or physical exam. Starting to sound familiar yet?
Myalgic encephalomyelitis was recognized as a neurological disorder by the World Health Organization (WHO) in 1969 based on evidence of central and peripheral nervous system damage in addition to muscular weakness. In addition to the neurological symptoms, doctors treating ME patients noted mental and cognitive changes similar to what we now call brain fog – impaired concentration, difficulty regulating emotion, wordfinding deficits. The primary literature from this period is rife with outdated terminology, but still physicians like Dr. A.M. Ramsay concluded that hysteria could not account for the epidemiology of this illness. A 1978 publication by Ramsay6 makes a troublingly prescient observation:
“Kendell (1967) has described in detail the case histories of two nurses aged 19 and 24 years. Both had bad psychiatric histories, and hysterical features were prominent in both their illnesses. But both had abnormal electromyograms and in one case paresis was unaffected by hypnosis and remained unchanged for 12 years while other symptoms disappeared. He had no hesitation in diagnosing both cases as instances of 'benign myalgic encephalomyelitis' and concluded with a most prophetic statement, 'the issue is important because other young women have been, and will continue to be, diagnosed as hysterical under similar circumstances with the resulting risk of their treatment being misdirected and their doctors' attitudes to them altered in unhelpful ways'. That is precisely what subsequently happened. [...] As a result of the widespread impression that they are 'neurotic' some have received scant sympathy or understanding from their doctors.”
Unfortunately, some of Dr. Ramsay’s colleagues disagreed with this viewpoint, publishing two papers in 1970 disputing a physiological basis for ME, suggesting that either patients or their doctors were inadvertently causing their own illnesses.7,8 This is one of the first papers to suggest that ME is a so-called “functional” syndrome, by which they mean symptoms are not being driven by a medical disease. Strike 1 for equating absence of evidence as evidence of absence.
. The text reads: “Summary: the reports of the 15 recorded outbreaks of benign myalgic encephalomyelitis have been reviewed and in one instance the original clinical data studied. We believe that a lot of these epidemics were psychosocial phenomena caused by one of two mechanisms, either mass hysteria on the part of the patients or altered medical perception of the community. We suggest that the name “myalgia nervosa” should be used for any future cases of functional disorder which present the same clinical picture.”")
The second paper, also published in the same issue of the British Medical Journal, took this idea several steps further, outlining what they considered to be “evidence” of mass hysteria:
 which reads: “Summary: from a re-analysis of the case notes of patients with Royal Free disease it is concluded that there is little evidence of an organic disease affecting the central nervous system and that epidemic hysteria is a much more likely explanation. The data which support this hypothesis are the high attack rate in females compared with males; the intensity of the malaise compared with the slight pyrexia; the presence of subjective features similar to those seen in a previous epidemic of hysterical overbreathing; the glove-and-stocking distribution of the anaesthesia; and normal findings in special investigations. Finally. a deliberate attempt by one of the authors to produce an electomyographic record similar to that reported in Royal Free disease was successful.”")
The most compelling evidence of a psychogenic origin according to these researchers? The high attack rate in women, of course! It is breathtaking to witness such blatant misogyny in published medical research. The authors also explicitly endorse the idea of absence of evidence being presented as evidence of absence, an especially damning conclusion considering how limited clinical testing was in 1970. The fact that one of the physicians was able to fake an electromyographic recording does not mean that the electrical recordings from patients were also falsified, although it’s amusing to imagine a pair of supposedly respectable doctors prodding themselves with electrodes, anxiously moving their limbs around until the recording looked just right, and then running to the BMJ to claim they had proven epidemic hysteria. The rationale here is hardly scientific but instead based on the characterization of complex female patients as neurotic, hysterical, and emotionally unstable.
The birth of chronic fatigue syndrome
In the 1980s there were three major outbreaks in the United States centered around Incline Village in Nevada, Chapel Hill in North Carolina, and the town of Lyndonville, New York. Doctors treating ME patients in these three areas independently contacted the Centers for Disease Control (CDC), worried that a novel pathogen might be causing illness in their communities; despite occurring during the same time period, clinicians at each site were never informed of the concurrent outbreaks in other areas. When the CDC finally dispatched a pair of investigators to Incline Village they appeared disinterested and reportedly “looked at test results, took some blood samples, and headed back.... without a word.” Perhaps inspired by Drs. McEvedy and Beard above, the CDC issued a report in 1986 referring to ME instead as a “syndrome of chronic fatigue” that would later inspire the rebranding of ME to chronic fatigue syndrome (CFS) in 1988 under the direction of the CDC.9
The clinicians from Incline Village suspected that the Epstein-Barr virus (EBV) was involved in ME pathology, the same virus that causes infectious mononucleosis and glandular fever. EBV is an example of a retrovirus, with the ability to insert its genome permanently into our cell’s DNA. While acute EBV infection usually resolves over a period of weeks or months, the virus remains dormant in our bodies forever and can become reactivated under extenuating circumstances, such as immunosuppression. This phenomenon is still quite difficult to study, but many of the Incline Village patients appeared to have chronic EBV with mononucleosis-like symptoms. Based on the hypothesis that latent EBV infection might be driving symptoms in ME patients, a clinical trial led by Dr. Stephen Straus10 administered the antiviral acyclovir to patients with “debilitating chronic fatigue of no clear cause.” Notably, the study included only 27 CFS patients and no healthy comparison group, instead comparing results within the same CFS patients randomized to receive control treatment or acyclovir. They measured antibody levels to various EBV proteins and other common viruses, as well as some basic blood panels. According to the authors none of these measurements yielded any significant differences between the treatment and control phases, although the primary data from these measurements were omitted from the final publication in the New England Journal of Medicine.10 Instead the authors noted that, mysteriously, patients reported better moods when they reported less severe symptoms.
. \"Although we could not identify a reliable laboratory marker of disease severity, we did find an association between the results of psychological tests and patients sense of well-being. Significant improvements in levels of anger, depression, and other mood states correlated with overall clinical improvement. These results indicate that affect plays an important part in the perception of illness severity in the chronic fatigue syndrome. Our findings are reminiscent of data showing that psychological factors contribute to one's vulnerability to delayed recovery from acute infections and are in accord with recent findings that a history of affective disorders is frequent among patients with the chronic fatigue syndrome.\"")
You read that correctly: a simple correlation between mood and symptom burden, alongside purported (not reported) negative results from blood testing following acyclovir treatment, was considered strong evidence of a psychogenic origin for CFS. Any patient who has experienced an energy-depleting illness can give you another explanation for such a correlation – being physically ill for long periods of time is inherently stressful, which will have clear negative impacts on mood. It should surprise nobody that patients reported less depression and anger when their symptom burdens were lighter, but when coupled with the assumption that a handful of blood tests can rule out every potential disease process, this “positive” correlation was unjustly claimed as evidence of psychological drivers. The lead author on this underpowered and poorly reported manuscript, Dr. Straus, would go on to become a top official at the National Institute of Allergy and Infectious Disease (NIAID) and would retain a sharp bias against CFS as a distinct clinical entity for the rest of his career, painting CFS patients as either neurotic or overly ambitious.
“Dr. Straus said his colleagues at the National Institutes of Health had demonstrated that many patients were psychologically ''different'' long before they developed the syndrome. He described some patients as having been anxious and depressed with various neurotic symptoms for years before becoming ill. In other cases, patients were motivated, dynamic, driven individuals who were functioning at peak levels when stricken. Some may be under an undue amount of stress trying to maintain busy lives.”
New York Times, 7/28/1988; Jane E. Brody
The unfortunate reality is that ME is a broad diagnosis by nature, likely encompassing many complex mechanisms that converge in similar systemic dysfunctions. Speaking from an analytical perspective, let’s recall that our ability to detect what we call statistically significant differences between groups depends on our ability to distinguish signal from noise. When we compare group means we are not just comparing two numbers but two distributions of numbers: whether or not the means are significantly different depends on their value and the overall pattern of data. Studies with small sample sizes are able to detect only the most dramatic differences, missing more subtle shifts or the emergence of distinct subgroups of responders. This is why preliminary studies are not considered to be definitive, and instead better viewed as starting points toward developing new hypotheses.
In regards to the acyclovir study, it was designed to test whether acyclovir could outperform control treatment in CFS patients and as such cannot draw any conclusions regarding causal mechanisms. I will also mention here that the control treatment was intravenous saline administered every 8 hours, and that expanding blood volume via intravenous fluids is now known to treat postural orthostatic tachycardia syndrome (POTS), an autonomic nervous system disorder that can present with hypovolemia.11 Dysautonomia is now included as a core diagnostic component of ME, meaning the “control” treatment in the acyclovir trial might have been treating these patients by simply increasing their blood volume; this could have been detected in the 1988 study with the inclusion of a control group receiving saline treatment.
In 1994 the CDC convened to create the now infamous Fukuda definition of CFS, a vague definition centered around chronic unexplained fatigued with at least 4 out of 8 nonspecific symptoms.12 To its credit this list of symptoms did include post-exertional malaise, but does not outright require PEM for diagnosis. This definition was a substantial deviation from the original disease of myalgic encephalomyelitis described by Dr. Ramsay, one of the most experienced doctors treating ME in the United States. Although most patients with the type of systemic disease described as ME would meet CFS criteria, the Fukuda criteria also captures patients who developed fatigue without any post-exertional symptoms or neuroimmune features. The Fukuda criteria require that all known causes of fatigue be ruled out, a proposition that sounds like it should reduce population heterogeneity but once again falls victim to the conflation of absence of evidence with evidence of absence. By framing CFS around a nonspecific symptom that is a feature of countless poorly understood disorders, the CDC appears to have intentionally morphed the original syndrome described by Ramsay and colleagues into a vague and heterogeneous syndrome that functions as a diagnosis of exclusion. Frustratingly, the Fukuda criteria continue to be used and cited in ME/CFS studies today despite the availability of newer criteria with strict inclusion requirements.
In fact, this appears to have been quite deliberate. In a 1994 letter obtained via the Freedom of Information Act, Dr. Straus reportedly wrote the following, emphasis added:9
“My own sense is that a few years of use [with the Fukuda definition] in the field will once again verify that there is no demonstrable or reproducible differences between individuals who meet the full CFS criteria and those who can be said to suffer Idiopathic Chronic Fatigue....
I predict that fatigue itself will remain the subject of considerable interest but the notion of a discrete form of fatiguing illness will evaporate. We would, then, be left with Chronic Fatigue that can be distinguished as Idiopathic or Secondary to an identifiable medical or psychiatric disorder. I consider this a desirable outcome.”
The biopsychosocial model of CFS
Throughout the 1990s and 2000s, the psychologization of ME began gaining real steam after the “biopsychosocial” model of health was popularized based on the theories of Dr. George Engel. When applied to CFS this is often termed chronic activity avoidance, a maladaptive response to an acute illness that results in physical deconditioning. According to this line of thought patients would try to return to their normal activity levels too quickly following an illness and experience a temporary worsening of symptoms, which caused them to erroneously conclude that exercise was making them sick. This would cause physical deconditioning, which would in turn decrease physical capacity and reinforce the misguided assumption that exertion is harming them. Of course, the cure for deconditioning brought on by activity avoidance is simple – you just need to gradually build up your exercise tolerance and go to therapy to address those “unhelpful cognitions” about your illness. This model of CFS as a behavioral disorder was promoted heavily by a group of psychiatrists in the United Kingdom, who received a large amount of funding in order to conduct a trial testing the efficacy of graded exercise therapy (GET) and cognitive-behavioral therapy (CBT) to treat CFS.
While deconditioning undoubtedly plays a role in exacerbating energy-limiting illnesses like ME, the BPS model assumes that altered perceptions around physical deconditioning are actually driving the disease (i.e. a ‘functional syndrome’) and not secondary to physiological abnormalities. It is true that graded exercise therapies can benefit some post-viral illness patients, particularly those who had very severe acute illnesses that required hospitalization and intensive care. The fundamental problem with this approach is not the assertion that some people could benefit from GET+CBT, but instead the assumption that all patients with medically unexplained fatigue had a behavioral or psychogenic disorder.
Patients who meet ME criteria experience a delayed and dramatic decline in functional status when they exceed their exertional capacities, and further studies would reveal dramatic changes in physical and metabolic capacities the day after a maximal exertion test that were not present in healthy or sedentary control subjects. This so-named post-exertional malaise (PEM) is debilitating and can lead to long-term deterioration if patients are repeatedly pushed past their exertional threshold and not given enough time or support to recover. In my personal experience, my friends with ME need support doing LESS activity and instead engaging in activity management and pacing strategies; these are people that have been largely house- or bedbound for months or years, who get excited and end up overdoing it on a “good” day and paying for it on the next. Prescribing an exercise program to these patients with the goal of gradually increasing physical stamina is a surefire way to instead progress toward increased severity and decreased function, especially when combined with CBT sessions explicitly intended to convince you that you aren’t really sick. Unfortunately, despite these limitations and contraindications, this GET+CBT combination remains the only intervention for CFS studied in a large clinical cohort.
The infamous PACE trial13 (Pacing, graded Activity, and Cognitive behaviour therapy; a randomized Evaluation) was conducted over 5 years and included 641 participants split into four different treatment groups: standard medical care, adaptive pacing therapy, GET+CBT, or adaptive pacing plus standard medical care. The major findings were published in 2011 in a paper that described CFS as a behavioral disorder in blatant language:
 CBT was done on the basis of the fear avoidance theory of chronic fatigue syndrome. This theory regards chronic fatigue syndrome as being reversible and that cognitive responses (fear of engaging in activity) and behavioural responses (avoidance of activity) are linked and interact with physiological processes to perpetuate fatigue. The aim of treatment was to change the behavioural and cognitive factors assumed to be responsible for perpetuation of the participant’s symptoms and disability. Therapeutic strategies guided participants to address unhelpful cognitions, including fears about symptoms or activity by testing them in behavioural experiments. These experiments consisted of establishing a baseline of activity and rest and a regular sleep pattern, and then making collaboratively planned gradual increases in both physical and mental activity. Furthermore, participants were helped to address social and emotional obstacles to improvement through problem-solving. Therapy manuals were based on manuals used in previous trials. CBT was delivered mainly by clinical psychologists and nurse therapists (webappendix p 1). Graded exercise therapy (GET) GET was done on the basis of deconditioning and exercise intolerance theories of chronic fatigue syndrome. These theories assume that the syndrome is perpetuated by reversible physiological changes of deconditioning and avoidance of activity. These changes result in the deconditioning being maintained and an increased perception of effort, leading to further inactivity. The aim of treatment was to help the participant gradually return to appropriate physical activities, reverse the deconditioning, and thereby reduce fatigue and disability. Therapeutic strategies consisted of establishment of a baseline of achievable exercise or physical activity, followed by a negotiated, incremental increase in the duration of time spent physically active. Target heart rate ranges were set when necessary to avoid overexertion, which eventually aimed at 30 min of light exercise five times a week. When this rate was achieved, the intensity and aerobic nature of the exercise was gradually increased, with participant feedback and mutual planning. The most commonly chosen exercise was walking. The therapy manual was based on that used in previous trials. GET was delivered by physiotherapists and one exercise physiologist (webappendix p 1).")
The primary outcome measures in the study were self-reported fatigue and self-reported physical function scores, and these subjective measurements showed moderate improvements in the GET+CBT group compared to standardized medical care and pacing. This positive result was not only hailed as a significant breakthrough for ME/CFS patients but also appeared to prove the behavioral and psychological nature of CFS. The GET+CBT approach was adopted across the world to treat the entity called CFS and the same band of psychiatrists continue to push their “fear avoidance” and deconditioning narratives on long COVID patients today
The debunking
The PACE trial quickly became controversial within the patient community considering the stark contrast between the reported results and the lived experiences of so many people with ME. Ethical issues were quickly identified by patient advocates, including failure to disclose ties to insurance companies during the informed consent process and erroneous media claims that CBT+GET can treat or cure CFS. Further scrutiny revealed a plethora of methodological and statistical flaws that challenge the interpretation of the published findings, which we will review here briefly. These have been extensively documented by journalist David Tuller and patient-researchers like Tom Kindlon, so please check out their linked pages for very detailed accounting.
The first issue arises with trial design: namely, behavioral interventions cannot be deployed with any patient or provider blinding. In other words, both patients and the clinicians interacting with patients are by necessity fully aware of their assigned intervention. This is already a major limitation in the context of controlled clinical trials but becomes more complicated by the reliance on subjective self-reported outcome measures. This combination alone renders the quality of any subsequent evidence low at best, but especially so when one group receives explicit coaching to discount their own symptoms. To try and account for some of these limitations, the study authors published the trial protocols well in advance including the criteria patients would have to meet in order to enroll in the trial.14 Their case definition of CFS did not require PEM but did require that patients showcase significantly impaired function as indicated by their SF-36 score, where lower scores equal lower functionality. Patients would have to score 60 or lower on this survey to enter the study, and would need to achieve a score of 75 or higher to be considered recovered. These thresholds were based on published data from the general population, in which healthy adults average a score of 85 and the 75 threshold fell approximately one standard deviation away from the mean. Setting these thresholds in advance is highly recommended to avoid biasing outcome measures in an unblinded trial.
In the publication of the PACE outcome data in the Lancet, the authors claim that approximately 60% of patients in the CBT+GET arm reached the recovery threshold by the conclusion of the year-long intervention. Digging deeper into the methodology section, the SF-36 entry criteria were relaxed from a score of 60/100 to a score of 65/100, reportedly to encourage recruitment. Inexplicably, the authors changed the threshold for recovery from 75/100 to 60/100 based on a post-hoc analysis of their dataset but failed to acknowledge or justify the changed criteria. This means that patients with an SF-36 score of 65 could theoretically enter the trial with a CFS diagnosis, deteriorate slightly for a final score of 61, and still be counted as having “recovered” according to the post-hoc analysis.

Further, the study did measure some objective outcomes, all of which failed to show anything resembling recovery. Movement tracking data and medical leave did not differ between groups, bringing into question whether participants in the CBT+GET group actually had decreased disability. The only significant difference was found in the 6-minute walk test in the GET group versus other groups, but the “improvement” in walking distance from 312m to 379m still leaves these patients severely limited in movement compared with people with class II heart failure (558m), cystic fibrosis (626m), and healthy elderly people (631m). Despite a very public campaign by the study authors to paint ME patients as unhinged critics, a FOIA request by Australian patient Alem Matthees finally succeeded in providing the community with deidentified data for secondary analyses. Before the data were released, the original study authors reanalyzed their own data and walked back their claim of a 60% recovery rate, instead concluding that approximately 20% of the GET+CBT group recovered (compared with 10% of controls). The independent reanalysis by Matthees and colleagues was even more devastating: when using the original proposed recovery thresholds, recovery rates were 4.4% for GET, 6.8% for CBT, and 3.1% of control arms.15 These low recovery rates were not significantly different between treatment groups. Despite these fundamental flaws and repeated calls for retraction from prominent researchers, the original study authors insist to this day that a combination of CBT and GET is the only evidence-based therapy for ME/CFS.
The evidence for metabolic dysfunction
Fortunately, by this time a handful of researchers across the US and Europe started thoroughly investigating potential physiological abnormalities in ME patients, including trying to characterize the experience of post-exertional malaise. In 2007, VanNess, Snell, and Stevens published the first paper to use two-day cardiopulmonary exercise testing (CPET) to quantify PEM.16 During a CPET, a patient performs upright exercise (cycling) that increases steadily in difficulty until they reach exhaustion. They are hooked up to all sorts of monitors and sensors during this test, so researchers can acquire detailed information about the patient’s respiration, oxygen consumption, peak heart rate, and more. When asked to perform a CPET on two consecutive days, healthy control participants were able to replicate or even exceed their day 1 performance when returning for day 2. ME patients did not appear significantly different from controls during day 1 of the study but showed significant deterioration in performance, oxygen consumption, and anaerobic threshold on day 2.

Follow-up studies by the same group of researchers replicated these findings in larger cohorts of CFS patients, concluding that diminished performance on the second CPET serves as objective evidence of post-exertional malaise. This phenotype appears unique to ME/CFS, as patients with other chronic illnesses like multiple sclerosis did not show impaired performance. A 2013 paper by Snell et al concludes:17
“The lack of any significant differences between groups for the first exercise test would appear to support a deconditioning hypothesis for CFS symptoms. However, the results from the second test indicated the presence of CFS-related postexertion fatigue. It might be concluded that a single exercise test is insufficient to reliably demonstrate functional impairment in people with CFS. A second test might be necessary to document the atypical recovery response and protracted fatigue possibly unique to CFS, which can severely limit productivity in the home and workplace.”
Other groups have shown major differences in the way ME/CFS patients respond to exertion. In a 2012 study, Light et al found major changes in gene expression in the blood of CFS patients following moderate exercise, and many of these genes are involved in sensory and autonomic signaling that correlated with self-reported measures of pain and fatigue.18

Further evidence supporting a biological driver of ME/CFS was published in 2014, in which Nakatomi et al used positron emission tomography (PET) to demonstrate ongoing neuroinflammation in ME/CFS patients versus healthy controls.19 This PET experiment utilized a radioactively labeled tracer that binds to a protein called TSPO (short for translocator protein). TSPO is expressed by cells in the brain called glial cells that provide critical support to neurons. Glial cells called astrocytes and microglia become activated in response to physiological stressors, and activated microglia in particular are known to dump inflammatory cytokines to promote local inflammation. These cells express the TSPO protein when they enter their “activated” state, meaning we can use PET technology to estimate the degree of neuroinflammation by measuring TSPO binding. Nakatomi et al found higher TSPO signal in several important brain regions in ME/CFS patients including the midbrain, pons, and thalamus – regions involved in autonomic regulation, motor coordination, motivation, and reward.

Interestingly, the authors also found that the degree of TSPO binding correlated very closely with self-reported measures of pain, cognitive impairment, and depression.

These studies all point toward a biological driver of ME/CFS. Due to the paltry funding situation, most of these studies are relatively small and there are some conflicting results, but the overall picture of the disorder paints it as neuroimmune response that fundamentally alters energy metabolism and impairs functional capacity. This leads us to 2015, the year in which the Institute of Medicine (IOM) released a report from their expert panels concluding that ME/CFS is a biological illness.20 In light of this report, the NIH announced it would fund an internal (intramural) study to “deeply characterize” ME that develops following an infection.2 While the IOM report clearly described ME/CFS as a disease rooted in an abnormal response to exertion, the language used by the NIH at this time still reflected a focus on the symptom of fatigue. Patients and advocates became concerned when the NIH announced this study would be led by Dr. Brian Walitt, whose research interests center around “perceptual illnesses”, or diseases driven by the mind. In a 2015 paper he co-authored about chemotherapy-induced cognitive dysfunction, the authors explicitly referred to CFS and fibromyalgia as “somatoform illnesses.”21 This pre-existing belief held by Dr. Walitt serves as a clear conflict of interest for the NIH’s largest study of ME/CFS, considering the lead author has already settled on a narrative for this illness that excludes the overt pathology the study claims to be interested in characterizing.
Over 8 years and 1 global pandemic later, the long-awaited study has finally been published. Despite a flurry of press releases and media articles lauding the paper as definitive proof of a biological driver in ME/CFS, patients are rightfully frustrated by the study’s focus on an oversimplified metric they call “effort preference” that was then plotted against seemingly every other variable studied to try and find some sort of correlation - as we will discuss Part II, this is not a particularly robust way to conduct analysis. The study only managed to enroll 17 participants, less than half of the originally proposed study size that was already criticized for being too small (the author list, on the other hand, has over 70 names). The manuscript inexplicably fails to mention post-exertional malaise outside of stating that it was a required symptom for enrollment; even their provided definition of ME/CFS neglects to convey that the disorder is centrally characterized by an abnormal response to exertion:
“Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is the commonly used term to describe a disorder of persistent and disabling fatigue, exercise intolerance, malaise, cognitive complaints, and physical symptoms with significant socioeconomic consequences. The physiological mechanism responsible for the persistence of fatigue and related symptoms has yet to be determined.”1
Notice here the framing once again suggests that ME/CFS is a disorder of nonspecific fatigue and other general symptoms that cannot be easily quantified by standard medical tests. ME/CFS is also presented as a socioeconomic problem, preventing people from working and consuming products and services. Despite the growing literature showing objective impairments in energy metabolism following a two-day maximum-performance exercise test, the study included only a single day of CPET testing which is known to be insufficient to distinguish ME/CFS patients from healthy controls. The way the data are presented and discussed in the text is often unclear and confusing, even for seasoned researchers (it’s me, I’m researchers). We will discuss these shortcomings and more in the next part of this series and break down the methods, highlighting both the problems and the potentially interesting results worth noting.
The same old song and dance
Before signing off, I wanted to take the opportunity to spotlight the way prominent COVID and long COVID (LC) minimizers have recycled the same talking points from the BPS brigade to target LC, the most recently described post-acute infection syndrome that impacts millions across the world. While public health agencies in both the US and the UK have since removed recommendations for GET+CBT therapies for ME/CFS, many of the same characters supporting the original PACE trial have strongly objected to the recategorization of ME/CFS as a biological illness. In a throwback to the original 1970 papers disputing a biological driver of ME, one particularly unpleasant commentary by Vogt and Garner22 tried to argue that disseminating medical information about long COVID actually threatened public health by making people too scared. This paper included one of the most disrespectful figures I have encountered in my time as an academic, making reference to Edvard Munch’s famous painting The Scream but in response to a newspaper discussing post-viral illness.

In the authors own words, they believe that much of the disability experienced by people with long COVID stems from inaccuracies in the way these patients are interpreting their own symptoms. This is just another repackaging of LC as a functional or somatic disorder, and another attempt to discredit the experiences of people who live with LC. The language they used to describe LC is eerily similar to the way Dr. Walitt described fibromyalgia and CFS as “disorders of perception”:
“This biomedical story, which links common symptoms to virally induced, permanent damage done to the body's parts is reductionist. We also find good reason to believe that it represents a pathogenic conceptualisation that plays into how people's brains interpret sensations. We know that our brains predict sensations, so symptoms are based on the brain's interpretation about how important or threatening something is to our integrity and wellbeing. Thus, the prevailing narrative about danger of permanent damage can lead to self‐perpetuating and self-validating cycle of symptom amplification.”
To be clear, no reliable experts are claiming that every instance of LC is the result of permanent bodily damage or harm. There is simply not enough information to prove or disprove such a dramatic assertion. On the other hand, we do know that infections can have long-term impacts in certain people; as an example, recent evidence has shown that the autoimmunity observed in multiple sclerosis is likely induced by EBV infection. Further, this so-called “analysis” fails to mention some critically important context: governmental and health authorities are already heavily minimizing the potential harms associated with COVID-19 infection. How can a “prevailing narrative about danger of permanent damage” cause a shadow pandemic while our press outlets, social media feeds, and government officials actively promote the narrative that COVID is no longer a threat to public health? It is curious how these authors seem more preoccupied with policing patient narratives rather than actually improving health outcomes.
References
1. Walitt, B. et al. Deep phenotyping of post-infectious myalgic encephalomyelitis/chronic fatigue syndrome. Nat Commun 15, 907 (2024).
2. NIH takes action to bolster research on Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. National Institutes of Health (NIH) https://www.nih.gov/news-events/news-releases/nih-takes-action-bolster-research-myalgic-encephalomyelitis/chronic-fatigue-syndrome (2015).
3. Marshall-Gradisnik, S. & Eaton-Fitch, N. Understanding myalgic encephalomyelitis. Science 377, 1150–1151 (2022).
4. Komaroff, A. L. & Lipkin, W. I. ME/CFS and Long COVID share similar symptoms and biological abnormalities: road map to the literature. Frontiers in Medicine 10, (2023).
5. Mirin, A. A., Dimmock, M. E. & Jason, L. A. Research update: The relation between ME/CFS disease burden and research funding in the USA. Work 66, 277–282 (2020).
6. Ramsay, A. M. ‘Epidemic neuromyasthenia’ 1955-1978. Postgrad Med J 54, 718–721 (1978).
7. McEvedy, C. P. & Beard, A. W. Royal Free Epidemic of 1955: A Reconsideration. Br Med J 1, 7–11 (1970).
8. McEvedy, C. P. & Beard, A. W. Concept of Benign Myalgic Encephalomyelitis. Br Med J 1, 11–15 (1970).
9. Dimmock, M. & Lazell-Fairman, M. Thirty Years of Disdain: How HHS and a Group of Psychiatrists Buried ME. (2015).
10. Straus, S. E. et al. Acyclovir Treatment of the Chronic Fatigue Syndrome. New England Journal of Medicine 319, 1692–1698 (1988).
11. Figueroa, R. A. et al. Acute volume loading and exercise capacity in postural tachycardia syndrome. Journal of Applied Physiology 117, 663–668 (2014).
12. Fukuda, K. et al. The Chronic Fatigue Syndrome: A Comprehensive Approach to Its Definition and Study. Ann Intern Med 121, 953–959 (1994).
13. White, P. D. et al. Comparison of adaptive pacing therapy, cognitive behaviour therapy, graded exercise therapy, and specialist medical care for chronic fatigue syndrome (PACE): a randomised trial. The Lancet 377, 823–836 (2011).
14. White, P. D. et al. Protocol for the PACE trial: A randomised controlled trial of adaptive pacing, cognitive behaviour therapy, and graded exercise as supplements to standardised specialist medical care versus standardised specialist medical care alone for patients with the chronic fatigue syndrome/myalgic encephalomyelitis or encephalopathy. BMC Neurology 7, 6 (2007).
15. Wilshire, C., Kindlon, T., Matthees, A. & McGrath, S. Can patients with chronic fatigue syndrome really recover after graded exercise or cognitive behavioural therapy? A critical commentary and preliminary re-analysis of the PACE trial. Fatigue: Biomedicine, Health & Behavior 5, 43–56 (2017).
16. VanNess, J. M., Snell, C. R. & Stevens, S. R. Diminished Cardiopulmonary Capacity During Post-Exertional Malaise. Journal Of Chronic Fatigue Syndrome 14, 77–85 (2007).
17. Snell, C. R., Stevens, S. R., Davenport, T. E. & Van Ness, J. M. Discriminative Validity of Metabolic and Workload Measurements for Identifying People With Chronic Fatigue Syndrome. Physical Therapy 93, 1484–1492 (2013).
18. Light, A. R., White, A. T., Hughen, R. W. & Light, K. C. Moderate Exercise Increases Expression for Sensory, Adrenergic, and Immune Genes in Chronic Fatigue Syndrome Patients But Not in Normal Subjects. The Journal of Pain 10, 1099–1112 (2009).
19. Nakatomi, Y. et al. Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An 11C-(R)-PK11195 PET Study. Journal of Nuclear Medicine 55, 945–950 (2014).
20. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. (National Academies Press, Washington, D.C., 2015). doi:10.17226/19012.
21. Wang, X.-M. et al. Chemobrain: A critical review and causal hypothesis of link between cytokines and epigenetic reprogramming associated with chemotherapy. Cytokine 72, 86–96 (2015).
22. Vogt, H. & Garner, P. ‘Long covid’ and how medical information is causing illness: A philosophical issue affecting public health. Journal of Evaluation in Clinical Practice n/a, (2023).
Thank you Janna. It’s appalling that we’re still having to talk about this. So much damage has been done via research conducted fraudulently in order to get a desired outcome.
Okay, I have a question about the PACE trial. I guess I should probably just look at it…but when you discuss their changing of the “recovered” threshold: did they not use an *improvement* or change in the SF-36 (or whatever it is called) to denote recovery? Or improvement? It doesn’t make sense that they would just give it a number when some could have gone from 65 to 61, like in your example, and they could call that person recovered. I am also curious to know if the de-identified data bore that out. I’m sure you don’t have that answer, but still curious. To be clear, I am NOT a fan of the PACE trial, this is just confusing to me.