
Deep dive: microclots in long COVID
Could persistent miniature blood clots drive long COVID symptoms?

Welcome to Long COVID Research Breakdown! For our inaugural post we will be reviewing a hot topic in long COVID research: microclots! If you’ve ever ventured into #longCOVID Twitter, you have probably seen chatter about these miniature blood clots that have been reported in patients with post-COVID illness. Curious about how these microclots form, where they might come from, and how they might cause post-viral symptoms? Read on to hear more about the research behind one of the major hypotheses for long COVID pathophysiology, and how I believe it complements other leading theories in the field!
TL/DR: the rundown
COVID-19 infections have been associated with coagulation deficits from the very beginning of the pandemic. It seems to do this by disrupting the endothelium, or the single layer of cells that line all of our blood vessels.
A group of researchers have found miniature blood clots in acute COVID-19 patients, as well as patients with long COVID. The clots are made up of a protein called fibrin that has taken on an anomalous, or amyloid, structure. Essentially, something changed the physical structure of fibrin to make it sticky, and prevent it from being broken down efficiently. These tiny clots aren’t large enough to block blood vessels, but can occlude capillaries, which is the major site of oxygen exchange for our tissues.
Researchers found that the S1 portion of the spike protein is sufficient to induce microclot formation in samples from healthy patients.
While both acute and long COVID patients have microclots, the clots in long COVID patients contained more inflammatory markers that can also contribute to altered coagulation processes.
Preliminary reports indicate 100% of long COVID patients have detectable microclots. Current research is being done to see if microclots can serve as a diagnostic biomarker for long COVID, and whether they correlate with certain symptoms or symptom severity.
Microclots could serve as a type of “underlying mechanism” in long COVID, as it intersects with several other lines of research including:
Metabolic dysfunction: persistent microclots that block capillaries could result in persistent cycles of oxygen deprivation, which eventually results in a state of oxidative stress. Metabolic dysfunction is heavily implicated in myalgic encephalomyelitis (ME), a post-viral condition characterized by severe fatigue and exertional intolerance. Many long COVID patients develop ME, and ME patients with no history of COVID infections also exhibited microclots.
Immune dysregulation: some studies have shown that white blood cells, which regulate immune responses in the blood, have metabolic and functional disruptions in long COVID patients. This would be in line with recent hypotheses that some long COVID cases may be the result of an underactive immune response, in which high viral loads or persistent viral pockets are more likely.
Endothelial disruption: continued metabolic dysfunction as well as dysregulated coagulation cascades can drive vascular damage, including loss of capillary density and resultant nerve damage.
Viral persistence: studies have shown that SARS-CoV-2 virus or antigen can persist in the tissue of some individuals for a long time, including those with long COVID. Persistent exposure to the spike protein could continue to drive microclot-related damage. However, this would also suggest that microclots are not the underlying cause of long COVID.
Vaccine injury: the S1 spike protein alone appears sufficient to induce microclot formation, at least in vitro. This indicates that encountering spike protein, whether through infection or vaccination, can drive vascular pathology. Some individuals may be predisposed to this effect and have adverse reactions to the high levels of spike protein induced in the mRNA vaccine. Importantly, COVID-19 infection remains a much higher risk factor for developing long COVID than vaccination.
The breakdown: SARS-CoV-2 as a vascular disease
From the onset of the COVID-19 pandemic we have known that SARS-CoV-2 infection can impact a very important part of our circulatory system called the endothelium, a single layer of cells that line our blood vessels that serves as a barrier between the blood in our system and the tough outer structure of the vessel. Despite being incredibly thin, this layer of cells plays a crucial role in promoting the interactions between different components of circulating blood, like platelet aggregation and blood fluidity.
 as it travels into the venous system via small venioles and larger veins. Endothelial cells line the vessels.")
The blood coagulation pathway walks a delicate balance between preventing clot formation in healthy tissue, promoting clotting during acute injury, and breaking down unnecessary clotting products. Early on in the pandemic, researchers and clinicians realized that patients with acute COVID-19 infection often presented with abnormal blood clotting, particularly surrounding the role of a protein called fibrinogen. Fibrinogen is short, soluble protein that is synthesized and released into our bloodstream by the liver. In response to an injury or vascular insult, the enzyme thrombin is released from the injury site and converts fibrinogen to fibrin, which is an alternative form of the protein that can essentially polymerize to form a blood clot. This system is an essential part of our defensive response, allowing us to heal from scrapes and cuts and even major wounds to prevent further blood loss. Traditional fibrin clots are broken down by the protein plasmin, which creates byproducts including the D-Dimer protein, which is often measured via blood test to check for recent clot development.

Strangely, patients with acute COVID-19 were presenting with paradoxical syndromes: some patients exhibited high clotting activity early on in the disease characterized by increased fibrinogen and other clotting factors, before progressing into a more severe disease state characterized by excessive bleeding. A group of researchers specializing in endothelial dysfunction and abnormal clotting - namely Drs. Resia Pretorius, Doug Kell and #TeamClots - hypothesize that both of these phenomena involve vascular dysfunction: upregulation of fibrin and fibrinogen can cause clot formation and damage the epithelium, while down-regulation can have the opposite effect. Our body has several homeostatic mechanisms to adapt to challenges like infections, but these responses can swing too severely and result in system-wide dysfunction. It was clear from the outset that SARS-CoV-2 could disrupt our circulatory system and our blood vessels, although whether or not this contributed to chronic post-acute sequelae was less clear.
SARS-CoV-2 and anomalous clotting
So what do fibrin and fibrinogen have to do with long COVID? Well, it turns out fibrin can actually play a big role in infection-associated vascular outcomes in another way: by undergoing conversion to an amyloid form. An amyloid protein is the peptide version of a giant, tangled knot of cables that are often found in the depths of my backpack: standard fibrin undergoes a physical shift in its structure to form a clumpy mess instead of the well-ordered sheets we expect to see during clot formation.
: Symptoms, Treatment & What it Is")
Many proteins are capable of entering an amyloid conformation – if the term sounds familiar, it is probably because amyloid forms of different proteins have been heavily implicated in diseases like Parkinson’s, Alzheimer’s, and certain forms of cancer. The core problem with amyloid-form proteins is their tendency toward aggregation, and they have the ability to self-propagate by inducing the same conformational change in protein structure to other proteins they encounter. Further, their abnormal structure makes them much more resistant to breakdown by the cellular pathways that normally regulate their levels in our bodies.
In a breakthrough study toward the end of 2020, Pretorius et al.1 demonstrated that amyloid forms of fibrin protein could be isolated from the blood of patients with acute COVID-19. The methods needed to visualize these clumps are simple and elegant: a blood sample is spun down in a centrifuge to separate the blood cells, platelets, and plasma serum. This “platelet-poor” plasma – so named due to the specialized separation method to rid the sample of the majority of clotting factors – can then be smeared on a slide and treated with a fluorescent dye that selectively labels proteins in amyloid conformation. The resulting signal can then be viewed and recorded with a fluorescent microscope, a standard piece of equipment available in many research labs. In healthy patients, there should be minimal signal present, indicating low or absent levels of misfolded fibrin. Notably, this study used blood samples collected from healthy control patients before the pandemic, so we know with certainty these individuals were never exposed to COVID-19. This paper would be the first of many describing abnormal fibrin clots in patients with COVID.


Interestingly, this paper also looked at samples from patients with Type 2 Diabetes Mellitus (T2DM) and found an increase in anomalous fibrin in those as well, although not to the same extent as COVID-19. This would suggest that fibrin microclots are not unique to COVID-19 or even to infections – other inflammatory or autoimmune conditions may also predispose an individual to abnormal clotting events. In fact, the T2DM samples were also collected before the COVID-19 pandemic (in 2018). Pretorius et al then devised a way to quantify the extent of microclot formation in blood samples using image processing techniques to calculate the percentage of each image occupied by amyloid protein signal. Increased clots, regardless of size or structure, should result in higher % of each image being attributable to anomalous fibrin. (For more information about how this methodology has grown over the past years, check out this Twitter thread by Dr. David Putrino)

Strikingly, it looked like the amount of amyloid clots present did not differ based on illness severity. If these amyloid fibrin clots can negatively impact our circulatory system and our endothelium, what could that indicate for post-COVID complications like embolisms or strokes, or about post-acute COVID symptoms like brain fog and exertional intolerance? Further, what is it about SARS-CoV-2 that makes it so adept at promoting microclots? Subsequent research has focused in on one main suspect: the S1 subunit of the spike protein.
SARS-CoV-2 spike protein: a potential culprit
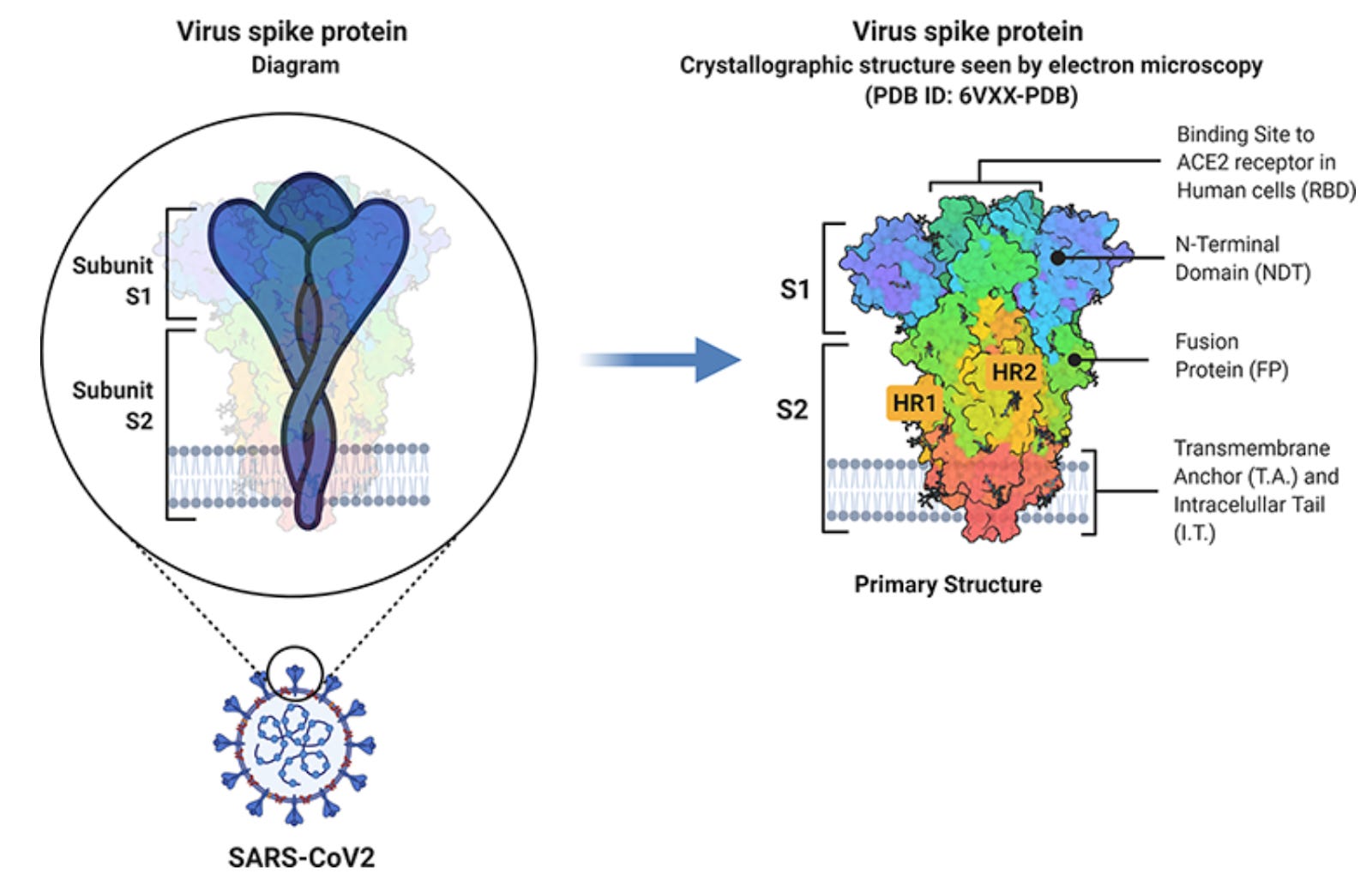
At a basic level, viral genomes are quite small and only contain a small number of protein-coding genes, mainly for proteins involved in the viral replication cycle: entering a cell, hijacking intracellular machinery, and multiplying. The spike protein protrudes off the membrane and contacts its surroundings, looking for an opportunity to bind to its host receptor, angiotensin-converting enzyme 2 (ACE2). ACE2 is expressed on the cell membranes of our respiratory and circulatory epithelium. The S1 subunit of the spike protein binds directly to ACE2, while the S2 portion anchors the protein to the membrane and facilitates entry into the cell. You have likely heard about spike protein – our mRNA vaccines encode versions of spike protein, and many blood tests have been developed to measure spike protein antibodies. This is because the spike protein is required for the virus to enter the cell and replicate, and thus represents a ubiquitous and accessible target for our immune system.* Interestingly, spike protein can also be shed into general circulation through infected cells releasing S1 particles. Could spike protein influence clot formation?
(*There are some recent exceptional discoveries of ACE2-independent cell entry in certain cell types, notably astrocytes in the brain2,3 and cardiac pericytes in the heart,4 both via a receptor called CD147. It’s clear we need a more complete picture of how SARS-CoV-2 can enter cells via alternative receptor interactions.)
In order to address this question, Grobbelaar et al (2021)5 isolated platelet-poor plasma from healthy donors and treated the samples with isolated S1 protein. They found that brief treatment with S1 spike was sufficient to induce anomalous clotting, with or without concurrent exposure to thrombin, an enzyme that induces clotting as a natural part of our coagulation cascade:

Similarly, spike protein was able to induce platelet activation – meaning our blood is “stickier” – as well as the ability to occlude microfluidic chambers pretreated with thrombin. The clots formed when spike protein was present were larger and more occlusive than healthy samples, and instead appeared more similar to samples from patients with acute COVID-19. From these studies, it appears that COVID-19, and the S1 spike protein in particular, may directly impact our circulatory system through dysregulation of the coagulation and fibrinolysis processes. This raises another question: could this play a role in long COVID?
In yet another study, Pretorius et al (2021)6 set out to measure coagulopathies in patients with long COVID. Using similar techniques from their other publications, they surveyed the blood of 11 long COVID patients and found various abnormalities from every patient – including one volunteer in which a prior blood sample was available for comparison.
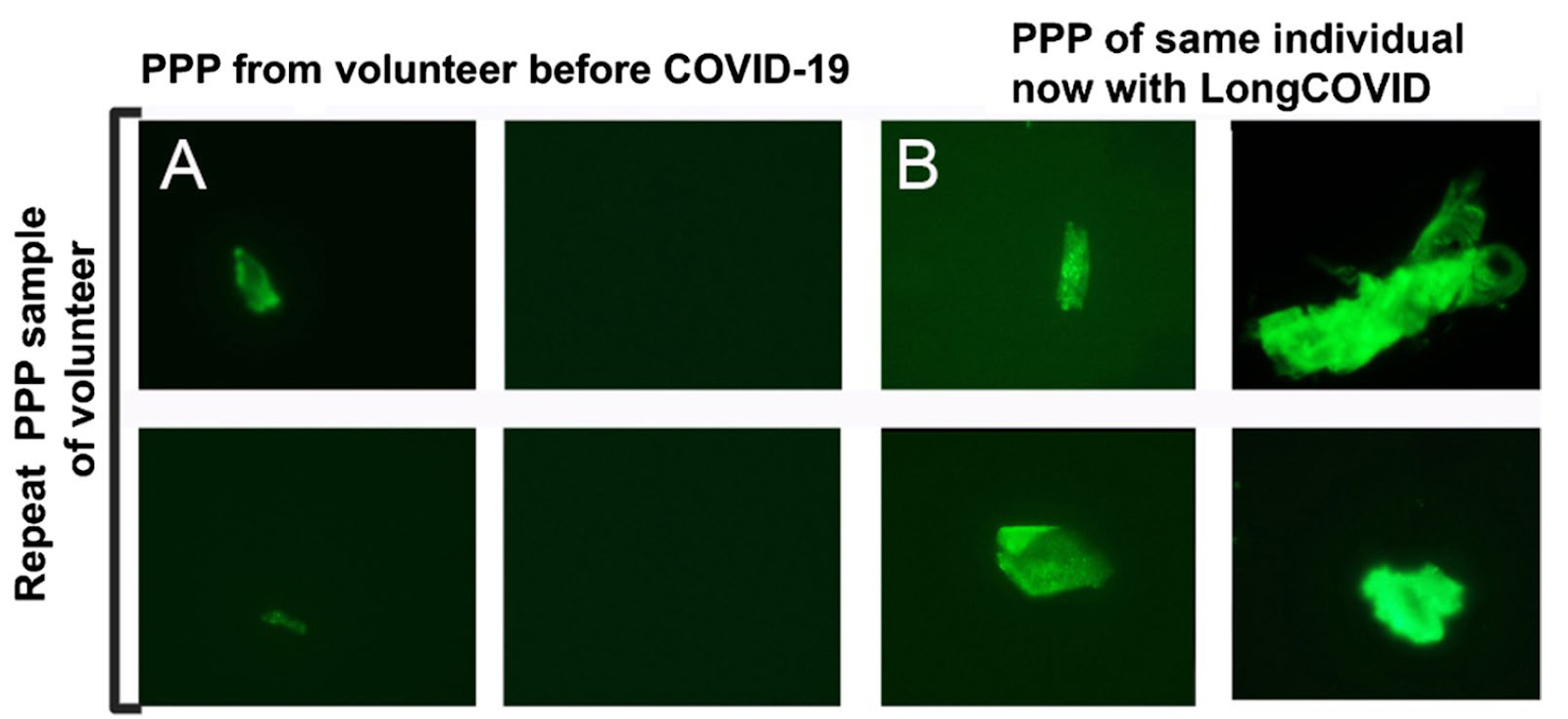
In addition to performing this analysis by microscopy, this study sought to find out what these clumps of anomalous fibrin might contain. In order to do this, the authors first took the platelet-poor plasma isolated from long COVID and healthy control patients – the same sample type visualized in the above figures – and exposed them to an enzyme that breaks down proteins. As a result, degradation-resistant proteins that were trapped in the fibrin-amyloid clot are now isolated in solution for further analysis.
Usually, when trying to study protein expression, you need a specifically developed antibody that reacts only to your protein of interest, which you can then quantify relative to control samples. Although offering good validity, this method is slow, requires higher sample volumes with additional protein targets, and importantly, requires that you already have a protein you’ve identified as interesting. Instead, the approach employed here – called proteomics– essentially breaks down proteins in a sample into chunks that are small enough to have distinct physical signatures at the chemical level on a mass spectrometer, but are also large enough to piece back together into their parent proteins using sophisticated computational approaches. The proteomics approach in Pretorius et al (2021) revealed that these microclots contained inflammatory proteins like plasminogen, α2 antiplasmin, von Willebrand Factor, and C-reactive protein. Importantly, several of these proteins were found in long COVID but were absent in acute COVID samples, suggesting an overlapping but distinct pathological state for acute versus post-acute COVID-19.

From this series of studies, we can begin to theorize about how SARS-CoV-2 infection may lead to both acute and post-acute consequences by shifting the balance of our coagulation and fibrinolysis system. SARS-CoV-2 infection, potentially through the spike protein, appears to trigger the formation of anomalous microclots that are resistant to degradation. An enzyme called plasmin is responsible for the breakdown of fibrin under normal conditions, and it circulates in the blood in a form called plasminogen that requires conversion into its active form. In order to activate, circulating fibrin must be present – and if lower amounts of non-anomalous fibrin are available in the blood due to long COVID, more fibrin will continue to accumulate into an amyloid formation instead of breaking down through the normal pathway. This could cause what we call a positive feedback loop, in which a shift in homeostasis drives itself further away from baseline through what is essentially self-propagation.
A potential biomarker for long COVID?
Since the publication of these studies, this so-named “microclot” hypothesis has become one of the most popular theories behind the pathophysiology of long COVID. These study leads and others have observed, for now anecdotally, that every long COVID patient tested to date has shown some degree of microclots. This poses an intriguing question: could the presence of microclots serve as a biomarker for long COVID, meaning could it be used to diagnose patients or measure the severity of their condition? Well, the answer to that is complicated.
It is important to note that, although these studies provide a very promising lead toward the underlying pathophysiology of long COVID, they all involve small sizes ranging from low double to low triple digits. While the technology used to visualize these clots is commonly available in research laboratories, they are not standard equipment in pathology labs or hospital settings. The method of testing is difficult to scale – using these methods, a skilled technician is required to prepare, mount, image, and individually analyze each sample. The process is fine for a few dozen samples at a time, but acquiring enough data to meaningfully determine whether these clots are diagnostic will take thousands of samples from both long COVID patients and healthy controls.
The process isn’t over once the samples are collected – afterwards, a subset of images from control and long COVID patients will be used to determine a standardized, computational approach that separates long COVID from control samples. Importantly, after this step the analysis needs to be repeated on novel samples to determine whether the effect has a high predictive ability, if it can be correlated with different symptom presentations, and if it can successfully differentiate between other inflammatory conditions like type 2 diabetes and ME/CFS. This is a big undertaking, but thankfully Dr. Resia Pretorius recently traveled to the United States to share her expertise on measuring and studying microclots with several groups leading this research – Drs. David Putrino (Mount Sinai), Akiko Iwasaki (Yale University), and Amy Proal (PolyBio Research Foundation), among others, are bringing the fight stateside. Drs. Putrino and Iwasaki are currently leading a clinical study seeking to do just this – I donated my blood late last year, and while I won’t know my own individual results, I’m pretty excited that my blood was some of the first in the country to be tested for microclots! For more, you can read this recent article featuring Dr. Putrino.
Microclots hypothesis – a potential unifying mechanism?
Just like we still don’t know whether microclots can serve as a definitive diagnostic tool for long COVID, we also don’t fully understand how – or if – these microclots contribute to long COVID pathophysiology. However, there are several compelling theories as to how these microclots could continue to drive pathological processes after an acute infection, which in my opinion makes the microclot hypothesis a compelling underlying mechanism that weaves together multiple theories. Please note that everything I discuss below is speculative, and while I have significant expertise in neuroscience and neurobiology, I am not an expert in this particular subject matter. If you are and have any feedback, I’d love to hear it (but please be kind)!
1. Metabolic dysfunction.
While these microclots are not large enough to occlude veins or arteries, they can disrupt the flow of blood through our capillaries – the tiny blood vessels that line all of our major organs, where our blood cells exchange oxygen to support our tissues. Our tissues need oxygen to survive – under normal conditions, our mitochondria generate energy using one of my favorite-named biochemical reactions, the electron transport chain. Choo choo!

If this is giving you flashbacks to high school biology, don’t worry – the details here aren’t important. Essentially, our mitochondria – the powerhouses of our cells – rely on a waterfall and a water wheel for their energy. However, this is a closed system and there is a limited source of water – and after a while, most of the water is going to end up on the bottom end of that waterfall. In order to keep things running smoothly, what do we do? We come up with a way to transport this water back to the top of the falls, in a way that uses less energy than it creates. Think about a water-powered pulley system – one that uses energy from the waterfall to carry buckets of water back up to the top, ensuring a constant supply of energy. This is the type of delicate balance our cells always strive for.
In the electron transport chain, our end product is still energy in the form of a molecule called adenosine triphosphate or ATP. ATP is made by an enzyme in our mitochondria called ATP synthase, which in our scenario is similar to the water wheel. Positively-charged protons in the form of H+ are pumped out of the inner matrix and into the intermembrane space, creating a charge gradient between the membranes – the intermembrane space is full of protons and is positively charged, while the matrix has few protons. The electron transport chain ensures that these H+ protons continue to be pumped back out of the matrix, to ensure ATP synthase has a steady supply of ions to keep making energy. The transport chain does this by grabbing negatively-charged electrons from molecules like NADH and FADH2 and passing them along the chain, until the final step where oxygen (O2) undergoes a reduction reaction to form 2 molecules of H2O, which we exhale in the form of water vapor.
What happens if the pulleys break, or the buckets begin to leak? Throw off the balance and soon enough, we’re out of water – in a pinch, we can rally a crew to haul buckets of water back to the top. This works well in an emergency, but it’s not exactly sustainable long-term. Similarly, during a state of low oxygen – hypoxia – our cells can use glucose instead of oxygen, a process called glycolysis or lactic acid fermentation. Glycolysis is fine in a pinch, but quickly results in a buildup of byproducts like lactic acid, a common culprit of sore muscles following a strenuous workout. Our glucose stores are not infinite, and glycolysis cannot sustain high energetic demand for very long. After a few trips up and down the hill, our friends don’t want to carry buckets anymore, so they go home.
Although this process typically occurs in muscles, it can also occur in other tissues following oxygen deprivation. So-called ischemic injuries often refer to conditions that develop during periods of disrupted blood supply, with strokes and heart attacks being extreme examples. One interesting thing I learned doing the research for this piece: the damaging effects of oxygen deprivation are not actually from a lack of oxygen, but instead from the reaction our oxygen-deprived system has when oxygen comes back.
Returning to our water wheel example – let’s say one of the mechanical bits powering the pulley system breaks. The item was custom-made, and it will take at least 1 year to get a replacement part. So the system stays abandoned for several years before the part finally arrives. If we immediately start pumping water back up the hill again without any inspection or maintenance, what happens? The buckets are all leaking, the tethers holding the pulleys are frayed, and the top of the waterfall is so dry the ground is cracked and damaged. We end up with more broken equipment and a mudslide – the damage from the drought outlasts the drought itself.
In our mitochondria, there’s a normal balance of protons being pumped out of the membrane in order to fuel ATP Synthase. During hypoxia, this charge gradient disappears – leading to a state of over-reduction in our electron transport chain, meaning there’s too many negatively-charged electrons with nowhere to go. When oxygen is finally reintroduced, these electrons can react violently with O2, forming peroxide (H2O2) instead of water, or even more severely, superoxide O2-.

Although some degree of oxidized molecules can be expected under baseline conditions, extended exposure to reactive oxygen species – the name given to these oxygen-containing molecules that react easily and violently with surrounding molecules – can lead to oxidative stress and the production of inflammatory molecules. As a result, COVID19 or spike protein-induced microclots could further self-propagate a cycle of inflammation by interfering with oxygen exchange at the molecular level, preventing our cells from producing enough energy to sustain themselves. It follows that microclots could potentially offer a unifying explanation for the variety of symptoms observed in long COVID patients across organ systems, which can mimic conditions of low oxygen saturation or poor perfusion (i.e. shortness of breath, exertional intolerance, orthostatic intolerance, fainting) without any corresponding evidence of hypoxia measured via pulse oximetry.
Further, mitochondrial dysregulation is heavily implicated in the energy-limiting illness myalgic encephalomyelitis, formerly called chronic fatigue syndrome (ME/CFS) – a condition that often manifests following a viral infection with a cardinal symptom of unrelenting fatigue that is not alleviated by rest. Approximately 50% of patients with long COVID also meet ME/CFS criteria, which includes a worsening of symptom severity following minimal physical or cognitive exertion.8 In fact, patients and clinicians alike quickly realized the two conditions share many characteristics, and may represent overlapping mechanisms of post-viral illness. Supporting this, ME/CFS patients without a history of COVID-19 infection also exhibit microclots and hyperactivated platelets similar to long COVID patients.9 Studies of ME/CFS patients have shown higher levels of oxidative stress and mitochondrial damage, specifically among circulating white blood cells.10–12 Similar reports have been published showing long COVID patients have impaired fatty acid metabolism and altered mitochondrial gene expression.13–16 While the exact mechanisms at play are still unknown, it’s clear that energy metabolism plays a fundamental role in ME/CFS and some presentations of long COVID, potentially due to oxidative stress induced by disrupted blood flow.
2. Immune dysregulation.
While it has long been assumed that long COVID has some sort of autoimmune component, recent evidence suggests some cases of post-COVID syndrome may result from an underactive immune response, in which persistent viral pockets, invasion of tissues, or reactivation of latent viruses becomes more likely.17–21 Coincidentally, SARS-CoV-2 has been shown to directly infect lymphocytes, a type of white blood cell in charge of our adaptive (i.e. “memory”) immune response. Persistent alterations in lymphocyte functions have been well documented in long COVID, including exhaustion of T cells and deficient dendritic cells, which function to flag down T cells and tell them where to go.17,22–32 Metabolic disruption could explain why these cells often appear “functionally exhausted” in long COVID.
3. Endothelial damage.
Accumulating evidence has shown long COVID patients can have extensive endotheliopathy, or disruption of the endothelium.33 Back at the very beginning of this post, we talked about the unique endothelium that lines our blood vessels and our respiratory tract – a single layer of cells that assist in maintaining the structural integrity of our blood vessels and form the site of important interactions in our blood, like responses to damage and immune challenges. Due to the sheer magnitude of tiny capillaries that supply blood to our tissues, endothelial disruption has the capacity to impact almost every part of our body. Several studies have shown elevated markers of endothelial injury in long COVID, as well as physical deformation of blood cells during acute infection.34–37 Common post-COVID symptoms like loss of taste and smell have been associated with microvascular injury in the olfactory nerve, where loss of vascular integrity leads to nerve degeneration.38 Similarly, long COVID patients have shown reduced capillary density, impaired oxygen extraction, and lung perfusion disturbances.39–41 I imagine this is connected to the high prevalence of autonomic nervous system dysfunction in long COVID and post viral illnesses more generally, but more research is needed to better understand this connection.
4. Viral persistence.
Several lines of evidence suggest that COVID-19 can form viral reservoirs or persistent pockets in areas of our bodies hard to reach by the immune system. In a recent autopsy study of patients who cleared the COVID-19 virus via nasal swab PCR but still progressed until death, SARS-CoV-2 proteins were detected in various tissues throughout the body, in one case 300 days post-infection.42,43 Persistent antigen has been detected in the brain, gut, lungs, liver, plasma, stool, and urine in PCR negative patients.43–49 It's still unclear whether these are active pockets of viral replication occurring in these tissues, if the virus enters some kind of latent cycle, or if viral molecules are somehow still being produced by formerly infected cells in the absence of replicating virus. However, some studies have shown persistent circulating spike protein in the blood of long COVID patients for at least 12 months.50 Viral persistence will get a deep dive post of its own soon, but continued production of and exposure to spike protein could explain the long-lasting nature of the microclots response. This would also suggest that microclots are a symptom instead of a cause – ridding the body of these clots through anticoagulant therapy or apheresis, in addition to having dangerous side effects, will not prevent more clots from forming without addressing the source of persistent spike.
5. Vaccine injury.
Vaccines are an important tool in our COVID-19 response, and they remain very effective at preventing hospitalization and death from COVID infections. However, there is a small but significant group of people who develop a post-COVID-like syndrome following vaccination, especially postural orthostatic tachycardia syndrome.51 Notably, the risk of developing this syndrome was 5x higher following COVID infection than vaccination – to me, these data suggest that exposure to high concentrations of spike protein via the mRNA vaccines may be sufficient to drive pathology in susceptible individuals, namely people who may be predisposed to autonomic dysfunction or coagulation deficits. In other words, the culprit behind these cases would seem to be spike protein, and not the vaccine itself - further suggesting that something about the spike protein in COVID-19 is able to do real damage to our bodies, whether it comes from an infection or from Pfizer. It is crucially important that we recognize these patients and their very real symptoms, and it will be further necessary to develop better vaccination strategies, including mechanisms to predict which patients are at high risk for developing these side effects.
So, are there any potential treatments?
(Note: I am a doctor but not THAT kind of doctor, this is not medical advice)
The popularity of the microclots hypothesis coincided with a subsequent rise in interest in experimental methods to target these clots. In a small group of long COVID patients with confirmed microclots, anticoagulant therapy was successful at reducing microclot density and improving symptoms.52 I had a difficult time finding information on over the counter blood thinners like aspirin, but it’s important to remember that traditional blood thinners A) have a myriad of potentially dangerous side effects and B) have only been clinically validated to treat large thromboses and not the breakdown-resistant microclots observed in long COVID. These therapies should be initiated and maintained under careful guidance from a medical professional. Similarly, apheresis clinics – in which your blood is filtered externally before being returned to your veins – have been popular among long COVID patients with the means, as these clinics typically charge tens of thousands of dollars per round of treatment. Again, it’s been difficult to find any concrete data on apheresis in long COVID, but the procedure comes with significant risks and is unlikely, in my opinion, to be curative. However, I will note that there is some evidence that removing certain autoantibodies via immunoadsorption apheresis may benefit certain patients clinically, but well powered studies will be needed to draw any definitive conclusions.
An alternative avenue many are pursuing are a group of supplements called fibrinolytics – as the name implies, these are enzymes that efficiently break down (lyse) fibrin. If you’ve heard about nattokinase, serrapeptase, or lumbrokinase, those are all various supplements that have these clot-busting properties without increased risks of bleeding. Many long COVID patients, including myself, have seen substantial improvements in some aspects of their symptoms shortly after beginning these supplements. If you would like to learn more, the Long COVID Pharmacist has a great in-depth post about safety, tolerability, efficacy, and dosage – I recommend following her newsletter too, she does great work!
That’s a wrap for today’s discussion on microclots! If you have any thoughts, ideas, or alternative hypotheses, let me know in the comments!
While you’re here, follow some of these awesome scientists and clinicians on Twitter!
Dr. Resia Pretorius, Distinguished Professor and Head of Department, Physiological Sciences, Stellenbosch University (South Africa)
Dr. Douglas Kell, Research Professor, Systems Biology, University of Liverpool; CSO Epoch Biodesign Ltd
Dr. David Putrino, Associate Professor and Director of Rehab Innovation at Mount Sinai (NYC)
Dr. Akiko Iwasaki, Sterling Professor of Immunobiology and Molecular Cellular, and Developmental Biology, Yale University; PI, Howard Hughes Medical Institute
Dr. Amy Proal, Microbiologist and CEO of PolyBio Research Foundation
References
1. Pretorius, E. et al. Prevalence of readily detected amyloid blood clots in ‘unclotted’ Type 2 Diabetes Mellitus and COVID-19 plasma: a preliminary report. Cardiovasc. Diabetol. 19, 193 (2020).
2. Crunfli, F. et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl. Acad. Sci. 119, e2200960119 (2022).
3. Andrews, M. G. et al. Tropism of SARS-CoV-2 for human cortical astrocytes. Proc. Natl. Acad. Sci. 119, e2122236119 (2022).
4. Avolio, E. et al. The SARS-CoV-2 Spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: a potential non-infective mechanism of COVID-19 microvascular disease. Clin. Sci. Lond. Engl. 1979 135, 2667–2689 (2021).
5. Grobbelaar, L. M. et al. SARS-CoV-2 spike protein S1 induces fibrin(ogen) resistant to fibrinolysis: implications for microclot formation in COVID-19. Biosci. Rep. 41, BSR20210611 (2021).
6. Pretorius, E. et al. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 20, 172 (2021).
7. Kell, D. B. & Pretorius, E. The potential role of ischaemia–reperfusion injury in chronic, relapsing diseases such as rheumatoid arthritis, Long COVID, and ME/CFS: evidence, mechanisms, and therapeutic implications. Biochem. J. 479, 1653–1708 (2022).
8. Davis, H. E., McCorkell, L., Vogel, J. M. & Topol, E. J. Long COVID: major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 1–14 (2023) doi:10.1038/s41579-022-00846-2.
9. Nunes, J. M., Kruger, A., Proal, A., Kell, D. B. & Pretorius, E. The Occurrence of Hyperactivated Platelets and Fibrinaloid Microclots in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Pharmaceuticals 15, 931 (2022).
10. Anderson, G. & Maes, M. Mitochondria and immunity in chronic fatigue syndrome. Prog. Neuropsychopharmacol. Biol. Psychiatry 103, 109976 (2020).
11. Schreiner, P. et al. Human Herpesvirus-6 Reactivation, Mitochondrial Fragmentation, and the Coordination of Antiviral and Metabolic Phenotypes in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. ImmunoHorizons 4, 201–215 (2020).
12. Wood, E., Hall, K. H. & Tate, W. Role of mitochondria, oxidative stress and the response to antioxidants in myalgic encephalomyelitis/chronic fatigue syndrome: A possible approach to SARS-CoV-2 ‘long-haulers’? Chronic Dis. Transl. Med. 7, 14–26 (2021).
13. Díaz-Resendiz, K. J. G. et al. Loss of mitochondrial membrane potential (ΔΨm) in leucocytes as post-COVID-19 sequelae. J. Leukoc. Biol. 112, 23–29 (2022).
14. Peluso, M. J. et al. SARS-CoV-2 and Mitochondrial Proteins in Neural-Derived Exosomes of COVID-19. Ann. Neurol. 91, 772–781 (2022).
15. Pozzi, A. COVID-19 and Mitochondrial Non-Coding RNAs: New Insights From Published Data. Front. Physiol. 12, (2022).
16. Galán, M. et al. Persistent Overactive Cytotoxic Immune Response in a Spanish Cohort of Individuals With Long-COVID: Identification of Diagnostic Biomarkers. Front. Immunol. 13, (2022).
17. Gaylis, N. B. et al. Reduced Cell Surface Levels of C-C Chemokine Receptor 5 and Immunosuppression in Long Coronavirus Disease 2019 Syndrome. Clin. Infect. Dis. 75, 1232–1234 (2022).
18. Hu, F. et al. A compromised specific humoral immune response against the SARS-CoV-2 receptor-binding domain is related to viral persistence and periodic shedding in the gastrointestinal tract. Cell. Mol. Immunol. 17, 1119–1125 (2020).
19. Jing, Y. et al. SARS-CoV-2 infection causes immunodeficiency in recovered patients by downregulating CD19 expression in B cells via enhancing B-cell metabolism. Signal Transduct. Target. Ther. 6, 1–13 (2021).
20. Peluso, M. J. et al. Impact of pre-existing chronic viral infection and reactivation on the development of long COVID. J. Clin. Invest. (2022) doi:10.1172/JCI163669.
21. Tian, W. et al. Immune suppression in the early stage of COVID-19 disease. Nat. Commun. 11, 5859 (2020).
22. Shen, X.-R. et al. ACE2-independent infection of T lymphocytes by SARS-CoV-2. Signal Transduct. Target. Ther. 7, 1–11 (2022).
23. Taeschler, P. et al. T-cell recovery and evidence of persistent immune activation 12 months after severe COVID-19. Allergy 77, 2468–2481 (2022).
24. Vijayakumar, B. et al. Immuno-proteomic profiling reveals aberrant immune cell regulation in the airways of individuals with ongoing post-COVID-19 respiratory disease. Immunity 55, 542-556.e5 (2022).
25. Diao, B. et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 11, (2020).
26. Glynne, P., Tahmasebi, N., Gant, V. & Gupta, R. Long COVID following mild SARS-CoV-2 infection: characteristic T cell alterations and response to antihistamines. J. Investig. Med. 70, 61–67 (2022).
27. Townsend, L. et al. Longitudinal Analysis of COVID-19 Patients Shows Age-Associated T Cell Changes Independent of Ongoing Ill-Health. Front. Immunol. 12, (2021).
28. Littlefield, K. M. et al. SARS-CoV-2-specific T cells associate with inflammation and reduced lung function in pulmonary post-acute sequalae of SARS-CoV-2. PLOS Pathog. 18, e1010359 (2022).
29. Peluso, M. J. et al. Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep. 36, (2021).
30. Pérez-Gómez, A. et al. Dendritic cell deficiencies persist seven months after SARS-CoV-2 infection. Cell. Mol. Immunol. 18, 2128–2139 (2021).
31. Phetsouphanh, C. et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 23, 210–216 (2022).
32. Chattopadhyay, P. et al. Single-cell multiomics revealed the dynamics of antigen presentation, immune response and T cell activation in the COVID-19 positive and recovered individuals. Front. Immunol. 13, (2022).
33. Ahamed, J. & Laurence, J. Long COVID endotheliopathy: hypothesized mechanisms and potential therapeutic approaches. J. Clin. Invest. 132, (2022).
34. Xu, S., Ilyas, I. & Weng, J. Endothelial dysfunction in COVID-19: an overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 1–15 (2022) doi:10.1038/s41401-022-00998-0.
35. Kubánková, M. et al. Physical phenotype of blood cells is altered in COVID-19. Biophys. J. 120, 2838–2847 (2021).
36. Patel, M. A. et al. Elevated vascular transformation blood biomarkers in Long-COVID indicate angiogenesis as a key pathophysiological mechanism. Mol. Med. 28, 122 (2022).
37. Charfeddine, S. et al. Long COVID 19 Syndrome: Is It Related to Microcirculation and Endothelial Dysfunction? Insights From TUN-EndCOV Study. Front. Cardiovasc. Med. 8, (2021).
38. Ho, C.-Y. et al. Postmortem Assessment of Olfactory Tissue Degeneration and Microvasculopathy in Patients With COVID-19. JAMA Neurol. 79, 544–553 (2022).
39. Osiaevi, I. et al. Persistent capillary rarefication in long COVID syndrome. Angiogenesis (2022) doi:10.1007/s10456-022-09850-9.
40. Heerdt, P. M., Shelley, B. & Singh, I. Impaired systemic oxygen extraction long after mild COVID-19: potential perioperative implications. Br. J. Anaesth. 128, e246–e249 (2022).
41. Yu, J. Z. et al. Lung perfusion disturbances in nonhospitalized post-COVID with dyspnea—A magnetic resonance imaging feasibility study. J. Intern. Med. 292, 941–956 (2022).
42. Bussani, R. et al. Persistent SARS-CoV-2 infection in patients seemingly recovered from COVID-19. J. Pathol. n/a,.
43. Stein, S. R. et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 1–6 (2022) doi:10.1038/s41586-022-05542-y.
44. Cheung, C. C. L. et al. Residual SARS-CoV-2 viral antigens detected in GI and hepatic tissues from five recovered patients with COVID-19. Gut 71, 226–229 (2022).
45. Menuchin-Lasowski, Y. et al. SARS-CoV-2 infects and replicates in photoreceptor and retinal ganglion cells of human retinal organoids. Stem Cell Rep. 17, 789–803 (2022).
46. Ceulemans, L. J. et al. Persistence of SARS-CoV-2 RNA in lung tissue after mild COVID-19. Lancet Respir. Med. 9, e78–e79 (2021).
47. Blagova, O. et al. Chronic biopsy proven post-COVID myoendocarditis with SARS-Cov-2 persistence and high level of antiheart antibodies. Clin. Cardiol. 45, 952–959 (2022).
48. Craddock, V. et al. Persistent Circulation of Soluble/EV-Linked Spike Protein and Viral RNA in Individuals with Post-Acute Sequelae of COVID-19. SSRN Scholarly Paper at https://doi.org/10.2139/ssrn.4186787 (2022).
49. Zollner, A. et al. Postacute COVID-19 is Characterized by Gut Viral Antigen Persistence in Inflammatory Bowel Diseases. Gastroenterology 163, 495-506.e8 (2022).
50. Swank, Z. et al. Persistent Circulating Severe Acute Respiratory Syndrome Coronavirus 2 Spike Is Associated With Post-acute Coronavirus Disease 2019 Sequelae. Clin. Infect. Dis. ciac722 (2022) doi:10.1093/cid/ciac722.
51. Kwan, A. C. et al. Apparent risks of postural orthostatic tachycardia syndrome diagnoses after COVID-19 vaccination and SARS-Cov-2 Infection. Nat. Cardiovasc. Res. 1–8 (2022) doi:10.1038/s44161-022-00177-8.
52. Pretorius, E. et al. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovasc. Diabetol. 21, 148 (2022).
Really wonderful summary and incite and access to detail that are spot on regarding long covid. From a treatment perspective: blocking at one or more steps on the general sequential path of: stop the virus>decrease inflammation>stop histamine and stop platelet activation>stop platelet aggregation>stop microclot>stop hypoxia>stop
Mitochondrial energy changes.
An approach as a general concept (not treating advice): truvada (tdf) or paxlovid for virus. Lower dose Metformin and/or higher dose Melatonin for inflammation and viral attachment. Antihistamines H1 and H2. Aspirin and plavix for antiplatelet and nattokinase for fibrin breakdown. Probiotics for gut health (and metformin helps to get more short chain fatty acid producers). More @doc4care on twitter. Thanks again for a phenomenal article!!
I am vax injured and in the listen study. Thank you for this information 💜❤️🩹