


The convoluted role of serotonin in infection-associated chronic illness
Could deficits in serotonin signaling explain cognitive dysfunction following viral infections?

TL/DR: The Rundown
A recent paper by Wong et al (2023) found decreased circulating serotonin levels were associated with long COVID, and used mouse models to show that viral inflammation can cause this effect by interfering with intestinal absorption and platelet destruction. In mouse models, viral infection had downstream effects on the vagus nerve and impacted novel object recognition.
Serotonin is synthesized from the amino acid tryptophan, which must be sourced from diet. However, tryptophan supplementation did not restore serotonin levels in virus-treated mice due to insufficient intestinal uptake.
Tryptophan is available over the counter as a nutritional supplement but has no evidence of therapeutic benefit for depression or sleep. People with myalgic encephalomyelitis (ME) should be particularly cautious with tryptophan supplementation, as side effects can exacerbate symptoms.
A theory by leading ME expert Dr. Ron Davis hypothesizes that common mutations in one of the genes involved in tryptophan metabolism, IDO2, could predispose individuals to develop ME. Loss of IDO2 function prevents the conversion of tryptophan to kynurenine when tryptophan concentrations are high, which could result in ME symptoms.
Selective Serotonin Reuptake Inhibitors (SSRIs) are commonly prescribed for depression and anxiety disorders. These drugs increase the concentration of serotonin within the synaptic cleft in the nervous system by blocking the serotonin transporter (SERT).
The majority of serotonin in the body is absorbed into circulation and taken up by platelets for storage via SERT. It doesn’t actually cross the blood-brain barrier and instead participates in platelet activation and aggregation. By blocking SERT, SSRIs prevent platelets from storing serotonin, which instead is degraded by monoamine oxidase.
Interestingly, the ability of SSRIs to reduce circulating serotonin levels may be implicated in their therapeutic effect, as people who respond well to SSRIs can be distinguished from non-responders by the magnitude of circulating serotonin suppression. People with depression are more likely to have cardiovascular health problems, and SSRIs might protect against this.
Despite serotonin’s known role in promoting platelet activation, Wong et al (2023) found that reduced serotonin levels also promoted platelet aggregation, an interesting distinction between major depression and viral infection.
The Breakdown
Welcome back to Long COVID Research Breakdown! I’m finally returning from hiatus to talk about the big-picture implications of a new blockbuster study1 published in Cell last month showing a mechanistic link between post-acute viral sequelae and reduced levels of circulating serotonin, a neurotransmitter linked to many physiological processes but most famously implicated in so-called “affective” disorders like generalized anxiety and major depression. Most laypeople are familiar with this molecule as the target of selective serotonin reuptake inhibitors (SSRIs), one of the most commonly prescribed classes of medication used to treat mood disorders: the classic monoamine theory of depression purports that reduced serotonin signaling in the brain underlies depressive phenotypes, which could explain why SSRIs and other similar medications can be effective treatments for depression.
Based on this understanding, one could incorrectly argue the recent finding supports a “biopsychosocial” or “psychogenic” basis to illnesses like long COVID because the mechanism appears to overlap with major depression and other mood disorders. However, this logic makes several assumptions that quickly begin to fall apart upon investigation, namely that SSRI therapy increases circulating serotonin levels (it does not!) and that increasing plasma serotonin levels through medications or nutritional supplements would have a therapeutic benefit in long COVID patients (unclear and potentially harmful).
We’re going to spend some time walking through these points and discussing what we actually know about the multifaceted role of serotonin in health and disease. My goal for this post is to convey an appreciation for the complexities surrounding receptor pharmacology and its connection to physiology and behavior. I argue that the Wong et al (2023) paper actually provides strong evidence against a psychological interpretation of long COVID and indeed concludes that viral infection produces measurable, distinct physiological changes in both mice and human volunteers with post-acute COVID syndrome (PACS). While the authors of the study were very careful to use language depicting PACS as an inherently physiological syndrome, such care does not always get translated to media coverage (or to your loved one that continuously recommends supplements and meditation as disease cures). Fear not - we’ll dive into this exciting topic together, starting with a recap of the serotonin paper that captured all of our attention.
Serotonin reduction in post-acute sequelae of viral infection
So what’s all the hype about anyways? For starters, the Wong et al (2023) paper was an incredibly thorough investigation that utilized both human volunteer cohorts and mouse models, allowing the authors to translate clinical findings into an animal model for more controlled mechanistic investigation. In fact, the paper integrated patient data from several long COVID patient cohorts after identifying serotonin dysregulation in acute COVID-19 patients, finding that circulating plasma serotonin levels in the post-acute phase could predict PASC status.

The paper goes on to corroborate this finding in several animal models: mice expressing a humanized ACE2 receptor and infected with wild-type (Wuhan Strain) SCoV2 and wild-type (non-modified) mice infected with vesicular stomatitis virus (VSV) and lymphocytic choriomeningitis virus (LCMV). VSV and LCMV are RNA-based viruses like SARS-CoV-2, and are well-established laboratory models of persistent viral infections. As an additional experimental group, the researchers injected mice with a compound called poly(I:C), which mimics nonspecific viral RNA and can stimulate an immune response. All of these rodent models had similar results: viral infection significantly decreased plasma serotonin levels, and further experiments show that this occurs through the suppression of amino acid absorption in the gut by viral inflammation.
The paper further explores the impact of viral infection on the brain using something called the novel object recognition test. In this behavioral paradigm, mice are placed in a neutral environment and provided access to a novel object - often simple laboratory tools like test tubes filled with sand, or large paper clips. After this initial session, the mouse is then presented with two objects: the same object from the first session, and a different novel object they have never seen before. Mice will naturally spend more time investigating the novel object during this test, which can be expressed as the novel object preference as a percentage of time.

As you might expect, VSV, LCMV, and poly(I:C) treatment reduced the amount of time the mice spent investigating the novel object:
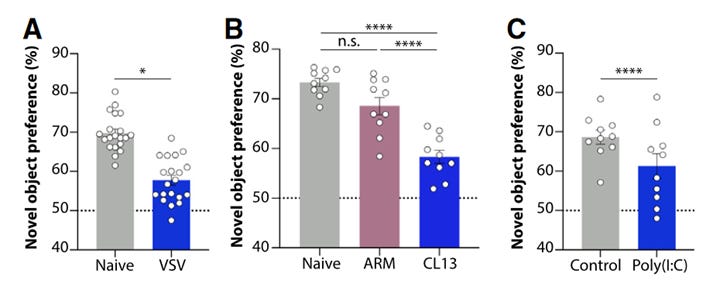
The authors are quick to call this a cognitive deficit, although as a (former) behavioral neuroscientist I disagree with that characterization. For starters, you can see in the graphs above (and in most of the expanded figures in the paper), the infected mice in the VSV, CL13, and poly(I:C) group spend less time investigating the novel object than the naive (untreated) mice. These statistical analyses relied on comparing each group of mice to each other to discern whether they are significantly different, as indicated by the asterisks. However, in this type of behavioral assay, there’s another set of statistics that should be run - namely, each individual group should also be compared with the threshold value of 50%. Although I can’t run these tests without the original data, it appears as though the statistically significant treatment groups above still spent the majority of the session interacting with the novel object - and spending less time investigating a novel object is not equivalent to failing to recognize the familiar object.
Why does this distinction matter? Behavioral studies in mice are useful for understanding the link between the brain and behavior, but the results are not simple to interpret. There could be many factors influencing a mouse’s behavior toward a novel object: ability to remember the familiar object, ability to recognize and recall the familiar object, ability to distinguish between novel and familiar objects, ability to regulate attentional processes to seek out novel objects, stimulation or suppression of locomotor activity, etc. Broadly speaking, behavioral experiments - especially those involving mice - are very sensitive and easily influenced by environmental factors. Mice and rats are prey animals, and consequently are prone to stress and anxiety during these procedures without proper acclimation and handling. This is why behavioral studies typically include a variety of behavioral tasks designed to measure similar aspects of the behavior of interest to further refine the phenotype. Regardless, the results here still clearly show a connection between viral infection and novel object responses, as poly(I:C) treatment led to a decrease in neuronal activation in the hippocampus and brainstem following the novel object recognition test.
Selective activation of the vagus nerve during poly(I:C) treatment restored the behavioral and neuronal responses to normal levels. Because the vagus nerve is directly activated by serotonin released from the gut, the authors conclude that reduced circulating serotonin leads to vagus nerve dysfunction in long COVID, a hypothesis consistent with many other reports.

The role of dietary tryptophan supplements
One obvious potential workaround for reduced gastrointestinal tryptophan absorption would be to supplement dietary tryptophan. However, this is unlikely to have any impact on circulating serotonin levels: Wong et al (2023) found that viral infection reduced the expression of genes necessary to absorb tryptophan from intestinal enterocyte cells (Slc6a19 and Ace2 below). This essentially creates a physical bottleneck that cannot be overcome simply through increasing dietary consumption.

On the other hand, the authors do show that supplementing a glycine-tryptophan dipeptide or 5-HTP in poly(I:C) treated mice did restore circulating serotonin levels - because these molecules can be absorbed in alternative ways that do not depend on the downregulated transporters. This is an important distinction, and highlights the complexities of amino acid synthesis and metabolism.

However promising this might sound, I believe that caution is warranted around dietary supplementation of tryptophan and other serotonin precursors, particularly in patients with myalgic encephalomyelitis (ME), formerly known as chronic fatigue syndrome. The cardinal symptom of ME is post-exertional malaise, or a dramatic worsening of symptoms following mild physical or cognitive exertion. The onset of ME typically follows some kind of immune system insult, with viral infection being the most common trigger. Although ME research has been systematically neglected by funding agencies over the past 40 years, a sizable body of work showcases dramatic metabolic and mitochondrial abnormalities in ME patients, suggesting a fundamental disruption in energy production. Approximately half of long COVID patients meet the diagnostic criteria for ME.
Although the definitive mechanisms driving ME are still unclear, many hypotheses center around fundamental dysregulation of energy metabolism at the cellular level. Mitochondria perform their powerhouse function through generating a molecule called adenosine triphosphate (ATP), which powers the enzymes that keep our cells running. What does tryptophan have to do with this? Tryptophan can also be converted into another bioactive metabolite, kynurenine. Kynurenine itself is then converted into either kynurenic acid or quinolinic acid. While kynurenic acid is broadly neuroprotective through limiting excitotoxicity, quinolinic acid can have the opposite impact - as well as depleting the molecule nicotinamide adenine dinucleotide (NAD), a necessary component of the mitochondrial electron transport chain that produces ATP.

If kynurenine sounds familiar, that’s because it was also highly dysregulated in COVID-19 patients in Wong et al (2023), but to the opposite degree: kynurenine and kynurenic acid levels were higher in acute COVID-19. However, figures from the supplemental materials show that kynurenine levels are not elevated in PASC patients and instead are lower compared with recovered controls. Tryptophan levels are also somewhat lower, which is consistent with decreased intestinal absorption.

The IDO metabolic trap hypothesis for ME
But there’s an additional layer of complexity surrounding kynurenine metabolism - the conversion from tryptophan to kynurenine involves two nearly identical enzymes, IDO1 and IDO2. When tryptophan levels are low, IDO1 takes the lead - but once they start to rise, its activity actually begins to decrease (an effect known as substrate inhibition, when the enzyme’s target molecule can also inhibit activity). This is where IDO2 comes in, favoring conditions with higher concentrations. There are several mutations in the IDO2 gene that can potentially decrease the enzyme’s activity, ranging from common (rs10109853, 0.418-0.487 allele frequency) to very rare (rs774492001, 0.000017-0.000020 allele frequency).2
With this information, Stanford geneticist and Human Genome Project alumni Dr. Ron Davis formulated the IDO metabolic trap hypothesis for ME, speculating that deleterious IDO2 mutations might function as a risk factor for ME by promoting a “metabolic trap” where high concentrations of tryptophan prevent kynurenine synthesis. This is supported by mathematical models of enzyme kinetics showing that a reduction of IDO2 activity by 90% fully prevents IDO activity across higher concentrations. Note here that IDO1 activity (flux) drops off starting at around 30 uM of tryptophan, and that the average tryptophan level even in tryptophan-depleted PASC subjects is around 45 uM (see above figure from Wong et al (2023)).

Under this theory, the decreased ability to convert tryptophan to kynurenine via IDO2 means that higher concentrations of cytosolic tryptophan will actually prevent kynurenine synthesis and its downstream metabolism. According to this hypothesis cells that regularly utilize tryptophan and its derivatives, like serotonergic neurons, enterochromaffin cells, and melatonin-producing pinealocytes, are most likely to be impacted.
Importantly, this is just a theory based on mathematical models - much more research will be needed to establish a definitive link between IDO2 and ME/CFS. However, I would still argue that this hypothesis warrants caution around the use of dietary tryptophan supplementation in people with ME, whether it was triggered by a COVID-19 infection or something else. Even in healthy individuals, tryptophan supplements can have detrimental side effects like nausea, cognitive difficulties, and heart palpitations - symptoms often present in ME, which could be further exacerbated by supplementation. There is no data to support the use of tryptophan supplementation for sleep or mood disorders, meaning they carry risks that are not offset by any potential benefits. The more direct serotonin precursor, 5-HTP, should theoretically bypass the IDO “trap” but there is still limited evidence to support its clinical use. Always discuss any new supplements with a knowledgeable clinician or specialist before taking them.
SSRI mechanisms and pharmacology
Chances are, you or somebody you know has probably taken a selective serotonin reuptake inhibitor (SSRI) for conditions like anxiety and depression. They are commonly understood to “boost” serotonin signaling - which is true, but fails to convey that the mechanism through which SSRIs increase serotonin is specific to the synapse. Let’s break this down.
Neurons in the brain are electrically active, but the majority of cell-to-cell communication in the nervous system occurs through what we call chemical transmission. Neurons have specialized, directional contact points called synapses where two neurons meet. The cells communicate by releasing neurotransmitters, which travel across the synapse to act on post-synaptic receptors. Chemical transmission between neurons gives rise to immense physiological complexity and forms the basis for principles like long-term potentiation of excitatory transmission following periods of high activity.
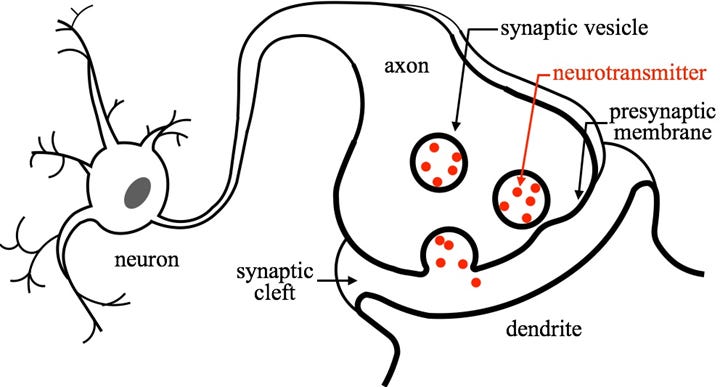
As you can imagine, neurotransmission is a tightly controlled process - various mechanisms are needed to ensure that the released neurotransmitters are quickly removed from the synaptic cleft to prevent diffusion to other sites. Neurotransmitter release is also energetically expensive, leading many neurons to engage in a recycling process where synaptic neurotransmitters are funneled back into the presynaptic neuron. This is where the term reuptake inhibitor comes from: SSRIs block the activity of the serotonin transporter (SERT), thereby increasing levels of serotonin only within the synapse.
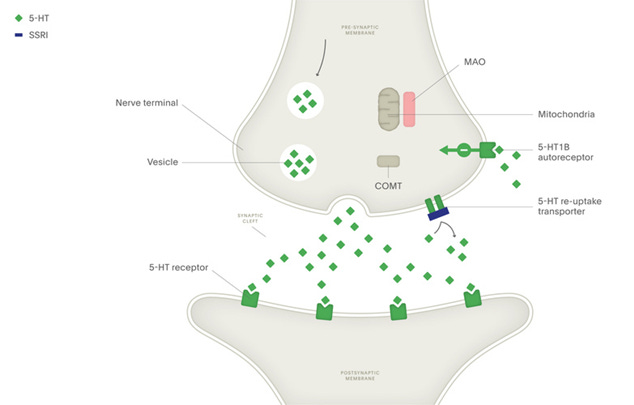
The SSRI medications are much more selective than previous generations of antidepressants like monoamine oxidase inhibitors and tricyclic antidepressants, which are no longer widely used due to the high degree of side effects. SSRIs are certainly better tolerated and more effective compared with these previous therapies, but at the clinical level their effect is typically modest, with many patients failing to respond to several different SSRIs. Furthermore, it takes weeks for the therapeutic effect of SSRI medications to develop - indicating the underlying mechanism is likely more complex than simply increasing synaptic serotonin. This is further complicated by the multifaceted nature of behaviorally defined disorders like depression, which likely encompass dozens of different mechanisms that converge in a similar behavioral phenotype.
This paper, alongside the widespread use of SSRI medications, raises a lot of questions about the relationship between serotonin and long COVID - namely, whether low plasma serotonin causes conditions like long COVID or are instead a symptom of underlying pathology. To this point, it’s important to note that serotonin levels at the postacute stage correlate with long COVID, but not serotonin levels during acute infection. Further, artificially depleting serum serotonin levels in mice did not induce inflammatory gene expression in the hippocampus, suggesting that serotonin depletion by itself is not sufficient to induce neuroinflammation. Considering the many studies that have shown persistent SARS-CoV-2 infection or viral gene expression in the gut mucosa, it follows that the serotonin deficiency and subsequent vagal dysfunction are downstream of disrupted gut absorption.
SSRIs and circulating serotonin levels
Due to the widespread understanding that SSRIs function by increasing serotonin concentration at the synapse, it’s tempting to assume that these drugs would similarly increase the concentration of serotonin in the blood. However, that’s not the case - circulating serotonin does not cross the blood-brain barrier, but instead can modulate nerve responses in the peripheral nervous system. Serum serotonin levels are actually maintained by platelets, which cannot synthesize serotonin but express the SERT transporter to vacuum up serotonin from the bloodstream before it's broken down by monoamine oxidase.
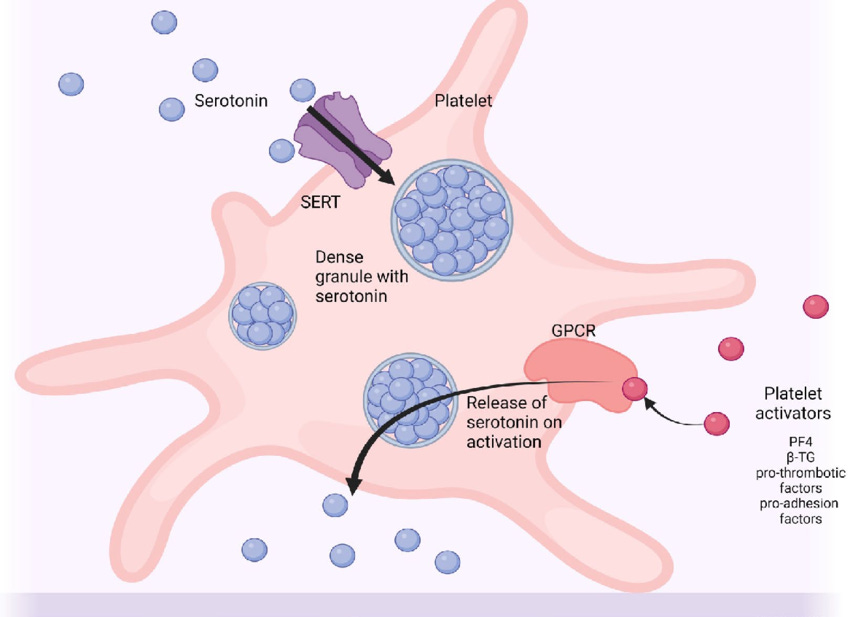
As you can see, blocking the SERT transporter with an SSRI will actually lead to lower platelet serotonin stores. This may seem counterintuitive at first, as decreased platelet serotonin would mean increased serum serotonin - but remember, free serotonin does not stick around for very long before encountering monoamine oxidase to break it down. Although initial treatment with SSRIs can increase plasma serotonin levels, chronic treatment causes sustained suppression of both intraplatelet and serum serotonin levels.3–8

In fact, studies show a greater drop in plasma serotonin is associated with positive response to SSRI therapy in depressed patients - implying the antidepressant mechanism involves reduced circulating serotonin, not an increase:

So how does this information tie into the Wong et al (2023) paper? Obviously, the effect of decreasing circulating serotonin levels is complex and multifaceted - in the case of SSRI therapy for depression or blood thinners for platelet depletion, reducing circulating serotonin levels might actually provide therapeutic benefits or correct deficits in serotonin signaling. On the other hand, infection by RNA viruses induces a host immune response that fundamentally disrupts our ability to synthesize serotonin from its dietary precursor, tryptophan. Following this logic, treating long COVID or other post-acute infection syndromes with SSRIs would likely exacerbate the circulating serotonin deficit and have reduced antidepressant efficacy.
A note on serotonin, platelets, and coagulation
Despite being best known as a neurotransmitter, serotonin has long been understood to act in the clotting cascade. Serotonin stores within platelets are released upon activation by pro-thrombotic factors, the downstream effects of which include vasoconstriction, inflammation, and conversion of fibrin to fibrinogen. Dysregulated and/or anomalous clotting has also been implicated in long COVID as well as serious cardiac sequelae following COVID-19, pointing toward another intriguing connection between the nervous and circulatory systems.9
Long before the COVID-19 pandemic began, there was a well-known association between major depressive disorder and poor cardiovascular health. There is also a significant body of research showing pronounced neuroinflammation in major depression, although these physiological signatures are often attributed to depressive behaviors despite lacking causal evidence. In addition to their more subjective antidepressant properties, SSRI therapy seems to have multifaceted benefits which can include reduced markers of inflammation and endothelial damage in depressed patients with coronary artery disease,10 reduced platelet aggregation,4,11 and reduced endothelial and platelet dysfunction following acute coronary events.12 Some studies even suggest that SSRIs can reduce heart attacks and mortality in the clinically depressed population,13,14 although there is some disagreement (i.e. Almuwaqqat 201915). Although direct evidence is lacking, these data are consistent with the idea that elevated circulating serotonin can have adverse cardiovascular consequences, which can be reversed through pharmacological blockade of the serotonin transporter.
Notably, platelets themselves can be depleted during viral inflammation as demonstrated by Wong et al (2023, Fig. 6B-D). Curiously, though, Wong et al demonstrated increased platelet activation following viral infection - unlike depressed patients with low serotonin following SSRI therapy, the low levels of serotonin induced by viral inflammation promotes platelet coagulation. What might explain this discrepancy? It’s hard for me to speculate as a non-domain expert in this area, and unfortunately the pro-coagulant properties of serotonin were not discussed in the Wong paper (no shade intended, these journals have strict page limits). Hopefully the authors of the Wong et al study are continuing their investigation of these phenomena. I think this is an excellent example highlighting the many layers of complexity surrounding neurotransmitter actions in the nervous system and beyond - and reiterates the point that caution is warranted when initiating therapies that impact serotonin signaling.
That’s a wrap for today’s post on serotonin in post-acute infection syndromes! What do you think? Have you had personal experience with these medications being prescribed for LC or ME? Let me know in the comments!
References:
1. Wong, A. C. et al. Serotonin reduction in post-acute sequelae of viral infection. Cell (2023) doi:10.1016/j.cell.2023.09.013.
2. Kashi, A. A., Davis, R. W. & Phair, R. D. The IDO Metabolic Trap Hypothesis for the Etiology of ME/CFS. Diagnostics 9, 82 (2019).
3. Abdelmalik, N. et al. Effect of the selective serotonin reuptake inhibitor paroxetine on platelet function is modified by a SLC6A4 serotonin transporter polymorphism. J. Thromb. Haemost. 6, 2168–2174 (2008).
4. Bismuth-Evenzal, Y. et al. Decreased serotonin content and reduced agonist-induced aggregation in platelets of patients chronically medicated with SSRI drugs. J. Affect. Disord. 136, 99–103 (2012).
5. Li, X. et al. Decreased platelet 5-hydroxytryptamin (5-HT) levels: a response to antidepressants. J. Affect. Disord. 187, 84–90 (2015).
6. Holck, A. et al. Plasma serotonin levels are associated with antidepressant response to SSRIs. J. Affect. Disord. 250, 65–70 (2019).
7. Honig, G., Jongsma, M. E., Hart, M. C. G. van der & Tecott, L. H. Chronic Citalopram Administration Causes a Sustained Suppression of Serotonin Synthesis in the Mouse Forebrain. PLOS ONE 4, e6797 (2009).
8. Reikvam, A.-G. et al. The effects of selective serotonin reuptake inhibitors on platelet function in whole blood and platelet concentrates. Platelets 23, 299–308 (2012).
9. Kell, D. B., Laubscher, G. J. & Pretorius, E. A central role for amyloid fibrin microclots in long COVID/PASC: origins and therapeutic implications. Biochem. J. 479, 537–559 (2022).
10. Pizzi, C. et al. Effects of Selective Serotonin Reuptake Inhibitor Therapy on Endothelial Function and Inflammatory Markers in Patients With Coronary Heart Disease. Clin. Pharmacol. Ther. 86, 527–532 (2009).
11. Serebruany, V. l. et al. Selective serotonin reuptake inhibitors yield additional antiplatelet protection in patients with congestive heart failure treated with antecedent aspirin. Eur. J. Heart Fail. 5, 517–521 (2003).
12. Serebruany, V. L. et al. Platelet/Endothelial Biomarkers in Depressed Patients Treated With the Selective Serotonin Reuptake Inhibitor Sertraline After Acute Coronary Events. Circulation 108, 939–944 (2003).
13. Scherrer, J. F. et al. Antidepressant Drug Compliance: Reduced Risk of MI and Mortality in Depressed Patients. Am. J. Med. 124, 318–324 (2011).
14. Fernandes, N. et al. The impact of SSRIs on mortality and cardiovascular events in patients with coronary artery disease and depression: systematic review and meta-analysis. Clin. Res. Cardiol. 110, 183–193 (2021).
15. Almuwaqqat, Z. et al. Association of Antidepressant Medication Type With the Incidence of Cardiovascular Disease in the ARIC Study. J. Am. Heart Assoc. 8, e012503 (2019).
This was fascinating thank you 🙏. It mirrors my experiences.
SARS-Cov-2 binds to Ace-2 receptors on platelets, causing a prothrombin estate and reduced platelet count.