


Lessons from low-dose naltrexone as a glial modulator to treat neuroinflammation
Is reducing the activation of microglia a viable treatment strategy for long COVID and myalgic encephalomyelitis?

Welcome to the Long COVID Research Breakdown! This month I’m covering low-dose naltrexone, a drug therapy gaining popularity as an off-label treatment for chronic inflammatory conditions like fibromyalgia, myalgic encephalomyelitis (former known as chronic fatigue syndrome), and most recently, long COVID. Although commonly known as a drug used to treat opioid use disorder, low-dose naltrexone appears to reduce signs of neuroinflammation through a totally different pharmacological mechanism, one with a very interesting history.
Before we dive in, one quick note – I am floored by all of the interest and enthusiasm on my first post on the microclots theory! I have lots of ideas for future long-form posts and deep dives and would love to hear from more of you about what kind topics and issues you’d like me to cover. Leave a comment below or head over to the “chat” page to start talking!
TL/DR: the rundown
Drugs exert their activity by acting on receptors on cells, which they can agonize (activate) or antagonize (block).
Naloxone and naltrexone are drugs developed to reverse acute opioid overdose and promote abstinence in people with opioid use disorder, respectively. These drugs are potent antagonists of opioid receptors, with naltrexone being much more easily absorbed by mouth than naloxone.
Several lines of work suggested that opioid drugs also interacted with an unknown receptor type, as administration of low doses of naltrexone (LDN) actually induced analgesia (pain relief) in rodents, an action more typical of an opioid agonist.
Initially, it was proposed that low-level blockade of opioid receptors by LDN would result in compensatory upregulation of endogenous endorphins. However, it was later discovered that naltrexone and naloxone also inhibit the toll-like receptor 4 (TLR4).
Stimulation of TLR4 results in the activation of microglia, the resident immune cell of the central nervous system. Once activated, microglia undergo a dramatic shift in morphology and release inflammatory molecules to stimulate T cell responses.
Several lines of work have shown that stress can potentiate microglial activation in response to a bacterial stressor, indicating that microglia may become “primed” to over-react to subsequent immune system challenges. This phenomenon, called neuroinflammation, is thought to play a role in many conditions including multiple sclerosis, Alzheimer’s disease, and ischemic injury.
As a drug, naltrexone actually comes in two forms, similarly to how we have a left a right hand. The drug we use clinically is called (-)-naltrexone, and antagonizes both opioid receptors and TLR4 receptors. This could explain why low doses exhibit anti-inflammatory processes that may become disguised by the opioid receptor blockade induced at higher doses.
The alternative form of the drugs, (+)-naltrexone and (+)-naloxone, selectively block TLR4 receptors without impacting opioid receptors. In animal models, (+)-naltrexone and/or (+)-naloxone have demonstrated significant neuroprotective benefits in many conditions. Although (+)-naltrexone seems like a promising drug for study, there is nowhere to source clinical-grade (+)-naltrexone. In the meantime, LDN can still help reduce symptoms associated with inflammation in long COVID, myalgic encephalomyelitis, multiple sclerosis, and more.
A new antibody therapy is in development that may be able to reduce microglial activation by preventing the activation of T cells, which would disrupt the viscous cycle between chronically activated microglia and functional T cell exhaustion. As a monoclonal antibody, this approach has the potential to reduce neuroinflammation with a much lower side effect profile. Phase 2a clinical trials are reported to begin later this year.
The breakdown: a tale of two receptors
In order to understand the different ways that naltrexone might work in long COVID and other complex illnesses, we will first need to review one of my favorite subjects: receptor pharmacology! You have probably heard about “receptors” in your body, especially when speaking about different medications and their interactions. But what is a receptor, actually?
At the simplest level, receptor proteins are the major way our cells communicate with each other and respond to their environments. Receptors are usually found on the surface of cells and other membranes and have specialized pockets, or binding sites, that recognize substrate molecules like neurotransmitters, antibodies, chemokines, and foreign particles. As a result of this substrate binding, a receptor undergoes a physical change – the protein sheets shift and the receptor will behave differently, for example by engaging downstream signaling processes that change cell activity. We can classify substrates into different categories based on where they bind and what they do: a substrate that directly activates the receptor and its downstream signaling is called an agonist, while a substrate that prevents the receptor from activating is called an antagonist.

All of our cells express receptors, and these receptors have natural activators often referred to as an endogenous agonist, meaning a receptor agonist that is produced naturally in our own bodies. As we learned more about receptors, researchers realized that drugs used medically or recreationally mediated their effects by acting within specific receptor families. This is why many receptor systems are named after drugs: nicotinic receptors utilize acetylcholine as their endogenous agonist, while opioid receptors naturally respond to a variety of endogenous endorphins. Receptor pharmacology quickly became a powerful approach to treating medical conditions and diseases – if disease is mediated by aberrant cell signaling, specific drugs could be applied to restore normal balance. For example, tachycardia and high blood pressure often result from high levels of the endogenous agonist epinephrine, which among other actions causes vasoconstriction through activation of β-adrenergic receptors in the circulatory system. Patients with tachycardia and high blood pressure are often prescribed beta blockers, a class of medications that antagonize β-adrenergic receptors to combat the effects of epinephrine.
Naltrexone as a therapy for opioid use disorder
Opioid drugs have been around long before receptor pharmacology was conceptualized. Humans in ancient cultures around the world consumed opium, plant material derived from the opium poppy that naturally contains around 12% morphine, to treat and manage pain. Opioid drugs are thought to treat pain by interrupting the relay between our different sensory systems, as the activation of mu opioid receptors (MORs) in the spinal cord and related structures prevent pain signals from being transmitted to the brain. However, these mu opioid receptors are also found in areas of our brain that process reward and motivation. In these regions mu opioid receptor agonists essentially remove the brakes from our dopamine system, which has the potential to rewire our reward system in a way that favors drug seeking – a hallmark of substance use disorders and the reason these drugs are classified as narcotics.
While opioid drugs remain the best treatment we currently have for acute pain the abuse potential of these treatments are high, and high doses of opioid agonist drugs can cause respiratory depression and death. These dangerous effects of the drug prompted the development of strategies to help patients in acute overdose or struggling with chronic opioid use disorder (OUD). Opioid receptor antagonists have since been discovered, with naloxone (Narcan) being frequently administered as a rescue therapy for acute opioid overdose. Naltrexone was first synthesized in the 1960s and was quickly classified as a potent antagonist at opioid receptors with almost identical properties to naloxone but was absorbed easily from the digestive tract, making it particularly promising as an oral therapy.
A major barrier to long-term recovery from OUD is navigating drug cues and cravings, which can lead to relapse. Naltrexone became the first FDA-approved therapy to treat OUD in 1984, prescribed in doses between 50-150 mg/day. It’s thought to do this by occupying and preventing signaling through MORs in the brain, which prevents the subjective “high” from opioid agonist drugs. Interestingly, naltrexone was more recently approved to treat alcohol use disorder (AUD) where it appears to reduce alcohol craving, although the mechanism of action for AUD is still unclear. However, these higher doses of naltrexone come with a major caveat: opioid therapy will no longer produce effective analgesia, complicating pain management.
Low-dose naltrexone: a pharmacological mystery
During its development in the 1980s, a few studies reported that low-dose naltrexone (LDN) had interesting pharmacological properties: despite being classified as a MOR antagonist, much lower (~1/10) doses of naltrexone appeared to act more like an agonist, inducing analgesia (pain relief) and boosting endorphin production in rats. While it appears as though LDN has been prescribed off-label for managing chronic conditions dating back to the 1980s, the first clinical study assessing LDN as a novel therapeutic was not published until 2007. Since then LDN – typically in doses of 4.5 mg/day, but can vary from 1.5-6 mg/day – has shown promise for treating several chronic neuro-immune disorders in small pilots studies for fibromyalgia, multiple sclerosis, inflammatory bowel disease, myalgic encephalomyelitis (ME, formerly known as chronic fatigue syndrome), and most recently, long COVID. However, these are not conditions that typically involve MOR signaling – and in fact, these effects aren’t seen with regular opioid agonists either. What is going on? LDN turns out to be a lot more complicated than anyone originally predicted!
Originally, the theory behind LDN’s mechanism of action still centered around MORs: it was proposed that transient, incomplete blockade of MORs by LDN caused the body to compensate by increasing the production of endorphins, which could in turn mediate long-term analgesia. However, a group of preclinical researchers had a different idea: what if naltrexone, at low doses, was acting on an entirely different type of receptor? That’s right, this whole spiel about opioid was all a red herring: LDN also appears to modulate a special type of cell called microglia, the resident immune cells of the central nervous system.
(Note: this discussion of LDN is not to be confused with ultra-low-dose naltrexone adjuvant approaches, in which microgram units of naltrexone administered alongside an opioid analgesic potentiates pain relief and reduces the overall dose administered, subsequently reducing the potential for side effects and dependence. Stay tuned for a bonus post on the pharmacological mechanism of ULDN, which is very interesting but not directly related to this topic.)
Microglia serve as a link between the nervous and immune systems
Microglia play a lot of important roles, including directing cell differentiation and connectivity during development. In the adult animal microglia typically hang out around the edges of the central nervous system in a dormant state, serving as sentinels for our immune system. When a threat is detected, these microglia spring into action: they become swollen, lose many of their branches, and begin synthesizing and releasing inflammatory molecules including cytokines and reactive oxygen species. In this state, microglia can also begin consuming the synapses that serve as relays between our neurons, a process called phagocytosis. While these processes are necessary in response to an injury or a pathogen encounter, persistent activation of microglia can damage and kill healthy cells in a process sometimes called neuroinflammation. Under high stress conditions, microglia are able to proliferate and migrate into the brain where they can continue driving immune responses.

Turns out, microglia play a role in something called sickness behaviors – a common set of behaviors observed across the animal kingdom during periods of acute illness. These include things we’ve all likely experienced such as reduced movement, food avoidance, weakness, fevers, and shaking. Sickness behaviors are correlated with high circulating levels of inflammatory cytokines, such as IL-1β and IL-6. This is an immune mediated response – when microglia and macrophages are exposed to certain stressors or antigens, they begin dumping these cytokines at the site of injury to recruit other necessary cells to complete the job. One of the most well-studied pathways in microglia is their activation through something called Toll-like receptor 4 (TLR4), which among things is activated by a common bacterial fragment called LPS. Agonism of TLR4 by LPS is sufficient to activate microglia and initiate cytokine release. These two different activation states have very distinct morphology and protein expression, making them relatively easy to discriminate – however as I like to say, nothing in biology is ever binary!
In 2003, Perry et al proposed a framework in which microglia and closely related macrophages can enter a third, “primed” state in which an additional trigger like an infection could generate an exaggerated and prolonged inflammatory response.2 The authors argue that chronic inflammation may underlie neurodegenerative processes seen in conditions like Alzheimer’s disease and multiple sclerosis, in which “primed” microglia are more apt to overreact to an infection or injury and set off inflammatory processes that ultimately cause damage. In 2007, Frank et al posited that microglial activation can be “primed” by prior stress to potentiate inflammatory responses in the brain.3 The authors subjected rats to a stressor, in this case a series of mild electrical shocks administered to the tail. 24 hours later, microglia were isolated from these rats and treated with LPS. The authors were thus able to directly examine the expression of different activity markers on microglia ex vivo and demonstrated that stress can indeed potentiate microglial activation in this model, resulting in increased levels of cytokine release compared to non-stressed (HCC, or “home cage control”) animals:
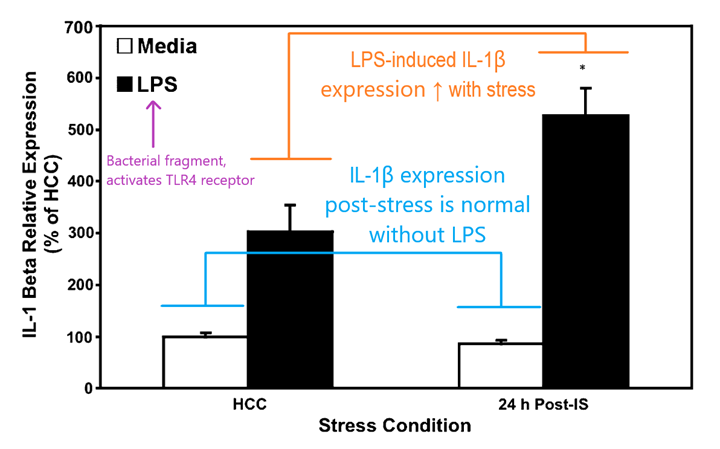
According to this framework, a combination of acute stress and immune challenge can result in a potentiated inflammatory response in the central nervous system versus an immune challenge alone. Given our understanding of what stress can do to the body and the brain this makes some intuitive sense. Microglia have only recently gained attention as an active component of neurological processes like pain, and our understanding of the way they act in the brain under different conditions is beginning to grow.
LDN suppresses microglial activation by blocking TLR4 receptors
Although opioids are best known for their ability to relieve pain via their activity at opioid receptors, they were more recently shown to alter the function of glial cells in the brain. Intriguingly, when morphine was administered to mice lacking opioid receptors it induced hyperalgesia (hypersensitivity to pain), implying that opioid drugs had physiological actions at non-opioid receptors. Subsequent studies have focused on the TLR4 receptor – originally discovered in fruit flies, TLRs recognize a variety of different exogenous and endogenous molecules like toxins, cellular debris, dying cells, and pathogens. As mentioned earlier, TLR4 is found on microglia and responds to a bacterial fragment called LPS, which acts as an agonist to activate the intracellular cascades responsible for cytokine production. Opioid agonists like morphine appear to have similar agonist properties at TLR4 receptors found in microglia.

Could opioid antagonists, like naltrexone, also antagonize (block) TLR4 receptors? The answer here requires us to go one step further in our understanding of chemistry, so let’s take a second to review a concept called chirality – essentially, “chiral objects” are asymmetrical in a way that a mirror image of the object cannot be superimposed onto it in the same plane. The word “chiral” comes from the Greek word for “hand”, which is one of the best methods of demonstrating this concept.
Hold your hands out in front of you, palm up. Your hands are mirror images – they look the same, only flipped in space. Now try to superimpose your hands, keeping your palms up. No matter what you do, your right and left hands are mirrors, but are not the same: they have a fundamentally different orientation in space.

Much like our hands, molecules can have different orientations in space, too. Even though we typically draw them as flat lines, chemical structures are 3-dimensional. Carbon, the building block of organic molecules, can form up to 4 bonds that make up a sort of cathedral structure. In chemistry we can include the relative position of bonds around a chiral carbon in our molecular diagrams by using dashed and bolded lines, like below. (If you were ever curious what organic chemistry is all about, it’s, well, a lot of this.)
Functionally, the carbon illustrated above is not chiral because all four of its bonds are identical. But when we start building complex organic molecules, we often encounter chiral carbons which contain 4 non-uniform bonds. There will thus arise two different “versions” of the molecule in question – much like we have a left and right hand, there is also a “left” and “right” molecule, which are referred to as stereoisomers and often designated as dextro- and levo-, or noted by a preceding (+) and (-). Naltrexone and naloxone, as well as many other opioid drugs, can exist as 2 stereoisomers as illustrated below:

As it turns out, the vast majority of studies to date on naltrexone and naloxone exclusively used the (-) enantiomer. Morphine and other opioid drugs are naturally found as the (-) enantiomer, and early testing in the 70s showed that (+)-morphine had little effect on opioid receptor responses or analgesia. You can think about it like trying to shake someone’s left hand with your right hand – all the right parts are there, but the pieces don’t fit together. However, (+)-morphine still induced hypersensitivity to pain in rats, indicating that (+)-morphine may have a non-opioid mechanism of action. These data would lay the foundation for the idea that (+) enantiomers may be acting through a distinct receptor subtype, but also precluded further development of (+)-naloxone and (+)-naltrexone for therapeutic potential. After all, when these drugs were developed, it was for the purpose of reversing the effects of opioid agonists. It would not make much sense to produce and administer two products when one has no known benefit.
However (+)-naloxone and (+)-naltrexone remained valuable research tools, and their original characterization in the 70s set the groundwork for the use of opioids as non-traditional immune system modulators. A group from the lab of Dr. Linda Watkins published a series of experiments in 2010 suggesting that both (+)- and (-)-naloxone and naltrexone inhibit TLR4 signaling in rats,4 and later work led by Dr. Watkins elegantly showed that (+)-naltrexone and (+)-naloxone directly inhibit TLR4 using microglia and macrophages from rats treated with LPS, a TLR4 agonist.5

It’s clear from this line of work that naltrexone has very interesting but complicated pharmacology, so let’s summarize: (-)-naltrexone blocks TLR4 receptors at lower doses and strongly blocks opioid receptors at high doses, while (+)-naltrexone has no effect on opioid receptors at any dose while still blocking TLR4 receptors. Under this framework it’s possible to explain the counterintuitive dose-response of (-)-naltrexone having different effects at low versus high dosage: at low doses TLR4 blockade is the dominant response, while higher doses can achieve opioid receptor blockade which can cause other cellular adaptations. It should also be noted that many of the commonly reported side effects of LDN, including difficulty sleeping, nausea, and decreased appetite, are also common side effects observed with opioid receptor blockade. We’ll circle back to that in a few paragraphs.
TLR4 inhibition in rodents reduces pain, ischemic injury
So what happens when we selectively inhibit TLR4 in rodent models using (+)-naltrexone? Subsequent studies by Dr. Watkins’ group have shown (+)-naloxone and (+)-naltrexone can reverse neuropathic pain, potentiate morphine-induced analgesia, and reduce microglial activation following nerve injury.4,6 It appears that TLR4 antagonism via (+)-naltrexone could provide an important brake on microglia-driven neuroinflammation in several different conditions.

Interestingly, (+)-naltrexone was shown to be protective in other situations. Survivors of cardiac arrest often exhibit signs of cognitive dysfunction or other memory deficits, which can be accompanied by neuronal death and damage in a region of the brain called the hippocampus. This inflammatory response typically occurs days after cardiac arrest and involves microglial activation. In a mouse model of cardiac arrest and cardiopulmonary resuscitation, systemically administered (+)-naltrexone decreased pro-inflammatory cytokine production and prevented cell death compared with controls receiving saline injections.7
Further study by another group administered intranasal (+)-naloxone to rats following a stroke and characterized microglial activation across the brain across various timepoints in recovery. One interesting finding in this paper: in the control group, microglia expressing activation markers were detected in the thalamus starting 1 week post-stroke, which eventually led to thalamic atrophy observed on day 112.8 This is surprising because the thalamus is far away from the origin site of the stroke injury, and suggests that any kind of ischemic injury can induce long-lasting activation of microglia in very distant brain regions. Activated microglia were observed along the neuronal projection fibers that connect the cortex to the thalamus, which could indicate these cells are traveling from the injury site to distal brain areas where they can drive secondary damage.

The good news is that intranasal (+)-naloxone administered daily post-stroke decreased both the resulting cell death in the cortex and in the distal thalamus region, suggesting that TLR4 blockade after ischemic injury is broadly neuroprotective by inhibiting microglia activation. Whether post-stroke naloxone administration represents an effective therapy for humans remains to be determined, but it seems clear that microglial activation plays a prominent role in ischemic injuries, stress-induced neuroimmune potentiation, and neuroinflammation.

LDN in the clinic
Throughout this period of discovery in basic preclinical research, a handful of clinical studies were published showing LDN can improve pain in some inflammatory conditions including fibromyalgia, a syndrome characterized by widespread pain and fatigue.9,10 Further supporting the hypothesis that LDN acts as an anti-inflammatory, a 2017 study showed 8 weeks of LDN treatment was associated with reduced circulating cytokines in fibromyalgia patients.11 Additional pilot studies have shown efficacy for LDN in treating Crohn’s disease and ME/CFS, conditions associated with widespread inflammation and cytokine release.12,13 Most recently, in a small study of adults with long COVID given LDN for 2 months; LDN treatment was associated with improvements in self-reported pain levels, energy, and quality of life.14 These trials typically used a dose of 4.5 mg/day, but doses up to 250 mg have been shown via metanalysis to carry very low risk of serious adverse events. Out of 11,194 participants in 89 controlled trials, only six “marginally significant adverse events” were reported, which were also mild in severity.15
It seems logical that LDN could represent a therapy option for long COVID: spike protein can directly drive microglial activation,16 several studies have shown elevated cytokine production in both severe acute COVID and long COVID,17–21 and ischemic-reperfusion injury driven by microclot formation is hypothesized to be a major driving mechanism behind long COVID22 (see my previous deep dive on the topic here). However, LDN supply both in the US and around the world is limited to (-)-naltrexone, the stereoisomer of naltrexone that can block both TLR4 and opioid receptors. Although considered to be an extremely safe and well-tolerated drug, patients have reported a variety of mild side effects from LDN therapy including insomnia, vivid dreaming, nausea, and loss of appetite. Notably, these are also side effects associated with opioid receptor blockade. Although the typical dosage of LDN is low enough that mu opioid receptors would only be partially occupied, it is also possible that conditions like long COVID may dysregulate endogenous opioid signaling in other ways that could make some individuals more sensitive to side effects. Why, then, are we still administering (-)-naltrexone when (+)-naltrexone could potentially reduce neuroinflammation with less side effects? The answer to that is complicated.
The TLR4-selective isomer (+)-naltrexone can be chemically synthesized in the lab, but to the best of my knowledge there are no available sources to dispense clinical-grade (+)-naltrexone that meet the necessary standards for human consumption set by the Food and Drug Administration. As a result studies using (+)-naltrexone and (+)-naloxone are limited to using animal models, and it’s unclear whether any major drug manufacturing company would be willing to front the time and money necessary to produce clinical-grade (+)-naltrexone. Although the efficacy of LDN in treating inflammatory conditions has support from many small pilot studies, there have been no large trials structured to determine optimal dosage and administration of (-)-naltrexone in these groups, not to mention (+)-naltrexone which could possibly be administered in higher doses. Naltrexone itself is also off-patent, which makes it unattractive to pharmaceutical companies seeking to maximize profits from their research and development efforts. However, one group has utilized the structure of (+)-naltrexone and the TLR4 receptor to engineer structurally similar compounds with ~75X more potent agonist activity versus (+)-naltrexone, which could represent a novel compound for drug development.23 Only time will tell whether a similar pharmacological approach will eventually land in the clinic.
Lessons from LDN: glial modulators for treating long COVID and inflammatory disorders
In my opinion, the biggest benefit from LDN is an important lesson in pharmacological discovery: how a drug prescribed as an off-label anti-inflammatory revealed novel insights into disease mechanisms and neuroinflammation. Although currently available LDN treatments are considered safe and appear to have a modest impact on symptom burden in many patients,14,24 the reliance on (-)-naltrexone means the maximal dosage will remain limited in order to avoid full opioid blockade. However, the discovery that (+)-naltrexone and (+)-naloxone selectively inhibit the TLR4 receptor in microglia and macrophages has driven the exciting hypothesis that neuroinflammation, mediated via persistently activated microglia, can be directly treated by blocking TLR4 activation with (+)-naltrexone. In an ideal world, I envision the characterization of alternative stereoisomeric ratios to determine the optimal levels of TLR4 and opioid receptor blockade for pain relief and inflammation, but these studies are unlikely to happen anytime soon due to the limitations discussed above.
However, there is some good news: the discovery that inhibition of microglia can reduce neuroinflammation recently led to the development of a potential alternative treatment for neuroinflammation – a monoclonal antibody that binds to a protein called CD3, which is found on a type of immune cell called T cells. Microglia and T cells are a type of dynamic duo: activated microglia stimulate the activation of T cells, which work together to ramp up the inflammation in chronic conditions.

Both severe acute COVID-19 and long COVID patients can exhibit T cell exhaustion,17,25–28 a state of immune dysfunction following chronic antigen stimulation. By binding to the CD3 receptor, Foralumab may be able to disrupt the vicious feedback loop between microglia and T cell activation following functional exhaustion and therefore indirectly reduce microglia activation. This drug is currently in testing phases for several conditions by the company Tiziana Life Sciences, which showed promise in a small study where patients with acute COVID-19 were given intranasal Foralumab daily for 10 days, then their T-cells were isolated and analyzed for gene expression using RNA sequencing. Foralumab treatment was associated with downregulation of inflammation-associated genes versus untreated patients, as well as in patients with multiple sclerosis.29 The treatment has also been piloted in healthy adults, in which no side effects were observed.30

Earlier in April, Tiziana announced their intention to pursue development of intranasal foralumab for the treatment of long COVID.31 Chairman of Tiziana’s Scientific Advisory Board Dr. Howard Weiner stated in the recent press release:
“In essence, intranasal foralumab modulates activated microglia while returning effector T cells to a naïve state. If this finding is replicated in patients with Long COVID, I believe that foralumab could represent a novel treatment for this devasting condition.”
Tiziana anticipates beginning a Phase 2a placebo-controlled trial later this year, where they will employ PET scans to non-invasively measure microglia activation following Foralumab treatment. Preliminary data from several patients with multiple sclerosis showing measurable changes in microglia activation following foralumab treatment was presented at a recent scientific meeting, but I have not been able to find another method to view this data, which presumably will be included in a forthcoming publication when more data has been collected. Regardless, if foralumab can indeed reduce neuroinflammation in long COVID and continues to be well tolerated, it will represent a huge breakthrough for the treatment of post-viral conditions and neuroinflammation.
That’s a wrap for today’s discussion on LDN! If you have any thoughts, ideas, or alternative hypotheses, let me know in the comments!
References
1. Woodburn, S. C., Bollinger, J. L. & Wohleb, E. S. The semantics of microglia activation: neuroinflammation, homeostasis, and stress. J. Neuroinflammation 18, 258 (2021).
2. Perry, V. H., Newman, T. A. & Cunningham, C. The impact of systemic infection on the progression of neurodegenerative disease. Nat. Rev. Neurosci. 4, 103–112 (2003).
3. Frank, M. G., Baratta, M. V., Sprunger, D. B., Watkins, L. R. & Maier, S. F. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain. Behav. Immun. 21, 47–59 (2007).
4. Hutchinson, M. R. et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain. Behav. Immun. 24, 83–95 (2010).
5. Wang, X. et al. Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br. J. Pharmacol. 173, 856–869 (2016).
6. Hutchinson, M. R. et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci. 28, 20–29 (2008).
7. Grace, P. M. et al. (+)-Naltrexone is neuroprotective and promotes alternative activation in the mouse hippocampus after cardiac arrest/cardiopulmonary resuscitation. Brain. Behav. Immun. 48, 115–122 (2015).
8. Anttila, J. E. et al. Post-stroke Intranasal (+)-Naloxone Delivery Reduces Microglial Activation and Improves Behavioral Recovery from Ischemic Injury. eNeuro 5, (2018).
9. Younger, J. & Mackey, S. Fibromyalgia Symptoms Are Reduced by Low-Dose Naltrexone: A Pilot Study. Pain Med. 10, 663–672 (2009).
10. Younger, J., Noor, N., McCue, R. & Mackey, S. Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum. 65, 529–538 (2013).
11. Parkitny, L. & Younger, J. Reduced Pro-Inflammatory Cytokines after Eight Weeks of Low-Dose Naltrexone for Fibromyalgia. Biomedicines 5, 16 (2017).
12. Lie, M. R. K. L. et al. Low dose Naltrexone for induction of remission in inflammatory bowel disease patients. J. Transl. Med. 16, 55 (2018).
13. Bolton, M. J., Chapman, B. P. & Marwijk, H. V. Low-dose naltrexone as a treatment for chronic fatigue syndrome. BMJ Case Rep. CP 13, e232502 (2020).
14. O’Kelly, B. et al. Safety and efficacy of low dose naltrexone in a long covid cohort; an interventional pre-post study. Brain Behav. Immun. - Health 24, 100485 (2022).
15. Bolton, M. et al. Serious adverse events reported in placebo randomised controlled trials of oral naltrexone: a systematic review and meta-analysis. BMC Med. 17, 10 (2019).
16. Albornoz, E. A. et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike protein. Mol. Psychiatry 1–16 (2022) doi:10.1038/s41380-022-01831-0.
17. Littlefield, K. M. et al. SARS-CoV-2-specific T cells associate with inflammation and reduced lung function in pulmonary post-acute sequalae of SARS-CoV-2. PLOS Pathog. 18, e1010359 (2022).
18. Hu, Z. et al. Early immune markers of clinical, virological, and immunological outcomes in patients with COVID-19: a multi-omics study. eLife 11, e77943 (2022).
19. Klein, J. et al. Distinguishing features of Long COVID identified through immune profiling. 2022.08.09.22278592 Preprint at https://doi.org/10.1101/2022.08.09.22278592 (2022).
20. Galán, M. et al. Persistent Overactive Cytotoxic Immune Response in a Spanish Cohort of Individuals With Long-COVID: Identification of Diagnostic Biomarkers. Front. Immunol. 13, (2022).
21. Peluso, M. J. et al. Markers of Immune Activation and Inflammation in Individuals With Postacute Sequelae of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Infect. Dis. 224, 1839–1848 (2021).
22. Kell, D. B. & Pretorius, E. The potential role of ischaemia–reperfusion injury in chronic, relapsing diseases such as rheumatoid arthritis, Long COVID, and ME/CFS: evidence, mechanisms, and therapeutic implications. Biochem. J. 479, 1653–1708 (2022).
23. Selfridge, B. R. et al. Structure–Activity Relationships of (+)-Naltrexone-Inspired Toll-like Receptor 4 (TLR4) Antagonists. J. Med. Chem. 58, 5038–5052 (2015).
24. LongCovidPharmD [@organichemusic]. To avoid any confusion, I am DELETING the posts with the medal emojis & ranking charts. I just wanted to explain why those posts are vanishing. However, I’m adding back the *individual* pie charts in this tweet & the next. Here’s ivabradine, LDN, & maraviroc. ⬇️ https://t.co/MOCkpqQtAY. Twitter
https://twitter.com/organichemusic/status/1567270707523903489
/photo/2.
25. Taeschler, P. et al. T-cell recovery and evidence of persistent immune activation 12 months after severe COVID-19. Allergy 77, 2468–2481 (2022).
26. Phetsouphanh, C. et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 23, 210–216 (2022).
27. Glynne, P., Tahmasebi, N., Gant, V. & Gupta, R. Long COVID following mild SARS-CoV-2 infection: characteristic T cell alterations and response to antihistamines. J. Investig. Med. 70, 61–67 (2022).
28. Diao, B. et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 11, (2020).
29. G. Moreira, T. et al. Nasal administration of anti-CD3 mAb (Foralumab) downregulates NKG7 and increases TGFB1 and GIMAP7 expression in T cells in subjects with COVID-19. Proc. Natl. Acad. Sci. 120, e2220272120 (2023).
30. Chitnis, T. et al. Nasal anti-CD3 monoclonal antibody (Foralumab) reduces PET microglial activation and blood inflammatory biomarkers in two patients with non-active secondary progressive MS. Mult. Scler. J. 839–840 (2022).
31. Tiziana Life Sciences to Pursue Development of Intranasal Foralumab for the Treatment of Long COVID. https://www.tizianalifesciences.com/news-item?s=2023-04-13-tiziana-life-sciences-to-pursue-development-of-intranasal-foralumab-for-the-treatment-of-long-covid (2023).
Header image from https://www.mdpi.com/2076-3425/10/3/159/htm
Thanks again Janna!
One thought I also have on LDN concerns the TRPM3-channel that seems to regulate Ca2+-influx into Natural Killer cells.
They seem dysregulated/overstimulated.
(+)-LDN seems to reverse this dysregulation in vitro in ME/CFS and LC pts.
https://pubmed.ncbi.nlm.nih.gov/31736966/
https://molmed.biomedcentral.com/articles/10.1186/s10020-022-00528-y
So mu-R antagonism seems needed in these instances.
Any thoughts on this?
How do I convince my physician to prescribe it??